
JANUVIA
These highlights do not include all the information needed to use JANUVIA safely and effectively. See full prescribing information for JANUVIA. JANUVIA (sitagliptin) tablets, for oral useInitial U.S. Approval: 2006
9d558f96-24a5-4788-8933-38256c17384b
HUMAN PRESCRIPTION DRUG LABEL
Apr 26, 2023
Cardinal Health 107, LLC
DUNS: 118546603
Products 1
Detailed information about drug products covered under this FDA approval, including NDC codes, dosage forms, ingredients, and administration routes.
sitagliptin
Product Details
FDA regulatory identification and product classification information
FDA Identifiers
Product Classification
Product Specifications
INGREDIENTS (12)
Drug Labeling Information
INDICATIONS & USAGE SECTION
1 INDICATIONS AND USAGE
JANUVIA® is indicated as an adjunct to diet and exercise to improve glycemic control in adults with type 2 diabetes mellitus.
Limitations of Use
JANUVIA should not be used in patients with type 1 diabetes.
JANUVIA has not been studied in patients with a history of pancreatitis. It is unknown whether patients with a history of pancreatitis are at increased risk for the development of pancreatitis while using JANUVIA. [See Warnings and Precautions (5.1).]
JANUVIA is a dipeptidyl peptidase-4 (DPP-4) inhibitor indicated as an adjunct to diet and exercise to improve glycemic control in adults with type 2 diabetes mellitus. (1)
Limitations of Use:
•
JANUVIA should not be used in patients with type 1 diabetes (1)
•
JANUVIA has not been studied in patients with a history of pancreatitis. (1, 5.1)
WARNINGS AND PRECAUTIONS SECTION
5 WARNINGS AND PRECAUTIONS
5.1 Pancreatitis
There have been postmarketing reports of acute pancreatitis, including fatal and non-fatal hemorrhagic or necrotizing pancreatitis, in patients taking JANUVIA. After initiation of JANUVIA, patients should be observed carefully for signs and symptoms of pancreatitis. If pancreatitis is suspected, JANUVIA should promptly be discontinued and appropriate management should be initiated. It is unknown whether patients with a history of pancreatitis are at increased risk for the development of pancreatitis while using JANUVIA.
5.2 Heart Failure
An association between dipeptidyl peptidase-4 (DPP-4) inhibitor treatment and heart failure has been observed in cardiovascular outcomes trials for two other members of the DPP-4 inhibitor class. These trials evaluated patients with type 2 diabetes mellitus and atherosclerotic cardiovascular disease.
Consider the risks and benefits of JANUVIA prior to initiating treatment in patients at risk for heart failure, such as those with a prior history of heart failure and a history of renal impairment, and observe these patients for signs and symptoms of heart failure during therapy. Advise patients of the characteristic symptoms of heart failure and to immediately report such symptoms. If heart failure develops, evaluate and manage according to current standards of care and consider discontinuation of JANUVIA.
5.3 Acute Renal Failure
There have been postmarketing reports of worsening renal function, including acute renal failure, sometimes requiring dialysis. A subset of these reports involved patients with renal impairment, some of whom were prescribed inappropriate doses of sitagliptin. A return to baseline levels of renal impairment has been observed with supportive treatment and discontinuation of potentially causative agents. Consideration can be given to cautiously reinitiating JANUVIA if another etiology is deemed likely to have precipitated the acute worsening of renal function.
Assessment of renal function is recommended prior to initiating JANUVIA and periodically thereafter. A dosage adjustment is recommended in patients with moderate or severe renal impairment and in patients with ESRD requiring hemodialysis or peritoneal dialysis. [See Dosage and Administration (2.2); Use in Specific Populations (8.6).]
5.4 Hypoglycemia with Concomitant Use with Insulin or Insulin Secretagogues
When JANUVIA was used in combination with insulin or insulin secretagogues (e.g., sulfonylurea), medications known to cause hypoglycemia, the incidence of hypoglycemia was increased over that of placebo used in combination with a sulfonylurea or with insulin. [See Adverse Reactions (6.1).] Therefore, a lower dose of sulfonylurea or insulin may be required to reduce the risk of hypoglycemia. [See Drug Interactions (7.1).]
5.5 Hypersensitivity Reactions
There have been postmarketing reports of serious hypersensitivity reactions in patients treated with JANUVIA. These reactions include anaphylaxis, angioedema, and exfoliative skin conditions including Stevens-Johnson syndrome. Onset of these reactions occurred within the first 3 months after initiation of treatment with JANUVIA, with some reports occurring after the first dose. If a hypersensitivity reaction is suspected, discontinue JANUVIA, assess for other potential causes for the event, and institute alternative treatment for diabetes. [See Adverse Reactions (6.2).]
Angioedema has also been reported with other DPP-4 inhibitors. Use caution in a patient with a history of angioedema with another DPP-4 inhibitor because it is unknown whether such patients will be predisposed to angioedema with JANUVIA.
5.6 Severe and Disabling Arthralgia
There have been postmarketing reports of severe and disabling arthralgia in patients taking DPP-4 inhibitors. The time to onset of symptoms following initiation of drug therapy varied from one day to years. Patients experienced relief of symptoms upon discontinuation of the medication. A subset of patients experienced a recurrence of symptoms when restarting the same drug or a different DPP-4 inhibitor. Consider DPP-4 inhibitors as a possible cause for severe joint pain and discontinue drug if appropriate.
5.7 Bullous Pemphigoid
Postmarketing cases of bullous pemphigoid requiring hospitalization have been reported with DPP-4 inhibitor use. In reported cases, patients typically recovered with topical or systemic immunosuppressive treatment and discontinuation of the DPP-4 inhibitor. Tell patients to report development of blisters or erosions while receiving JANUVIA. If bullous pemphigoid is suspected, JANUVIA should be discontinued and referral to a dermatologist should be considered for diagnosis and appropriate treatment.
•
Pancreatitis: There have been postmarketing reports of acute pancreatitis, including fatal and non-fatal hemorrhagic or necrotizing pancreatitis. If pancreatitis is suspected, promptly discontinue JANUVIA. (5.1)
•
Heart failure: Heart failure has been observed with two other members of the DPP-4 inhibitor class. Consider risks and benefits of JANUVIA in patients who have known risk factors for heart failure. Monitor patients for signs and symptoms. (5.2)
•
Acute Renal Failure: Has been reported postmarketing, sometimes requiring dialysis. Assessment of renal function is recommended prior to initiating JANUVIA and periodically thereafter. (5.3)
•
Hypoglycemia with Concomitant Use with Insulin or Insulin Secretagogues: Increased risk of hypoglycemia when used in combination with insulin and/or an insulin secretagogue. Lower dose of insulin or insulin secretagogue may be required. (5.4, 7.1)
•
Hypersensitivity Reactions: There have been postmarketing reports of serious allergic and hypersensitivity reactions in patients treated with JANUVIA such as anaphylaxis, angioedema, and exfoliative skin conditions including Stevens-Johnson syndrome. Promptly stop JANUVIA, assess for other potential causes, institute appropriate monitoring and treatment. (5.5, 6.2)
•
Severe and Disabling Arthralgia: Has been reported in patients taking DPP-4 inhibitors. Consider as a possible cause for severe joint pain and discontinue drug if appropriate. (5.6)
•
Bullous Pemphigoid: There have been postmarketing reports requiring hospitalization in patients taking DPP-4 inhibitors. Tell patients to report development of blisters or erosions. If bullous pemphigoid is suspected, discontinue JANUVIA. (5.7)
DOSAGE & ADMINISTRATION SECTION
2 DOSAGE AND ADMINISTRATION
2.1 Recommended Dosing
The recommended dose of JANUVIA is 100 mg once daily. JANUVIA can be taken with or without food.
2.2 Recommendations for Use in Renal Impairment
Assess renal function prior to initiation of JANUVIA and periodically thereafter.
For patients with an estimated glomerular filtration rate [eGFR] greater than or equal to 45 mL/min/1.73 m2 to less than 90 mL/min/1.73 m2, no dosage adjustment for JANUVIA is required.
For patients with moderate renal impairment (eGFR greater than or equal to 30 mL/min/1.73 m2 to less than 45 mL/min/1.73 m2), the dose of JANUVIA is 50 mg once daily.
For patients with severe renal impairment (eGFR less than 30 mL/min/1.73 m2) or with end-stage renal disease (ESRD) requiring hemodialysis or peritoneal dialysis, the dose of JANUVIA is 25 mg once daily. JANUVIA may be administered without regard to the timing of dialysis.
The recommended dose of JANUVIA is 100 mg once daily. JANUVIA can be taken with or without food. (2.1)
Dosage adjustment is recommended for patients with eGFR less than 45 mL/min/1.73 m2. (2.2)
Dosage Adjustment in Patients with Renal Impairment (2.2)|
eGFR greater than or equal to 30 mL/min/1.73 m2 to less than 45 mL/min/1.73 m2 |
eGFR less than 30 mL/min/1.73 m2 (including patients with end stage renal disease [ESRD] on dialysis) |
|
50 mg once daily |
25 mg once daily |
DESCRIPTION SECTION
11 DESCRIPTION
JANUVIA Tablets contain sitagliptin phosphate, an orally-active inhibitor of the dipeptidyl peptidase-4 (DPP-4) enzyme.
Sitagliptin phosphate monohydrate is described chemically as 7-[(3R)-3-amino-1-oxo-4-(2,4,5-trifluorophenyl)butyl]-5,6,7,8-tetrahydro-3-(trifluoromethyl)-1,2,4-triazolo[4,3-a]pyrazine phosphate (1:1) monohydrate.
The empirical formula is C16H15F6N5O•H3PO4•H2O and the molecular weight is 523.32. The structural formula is:
|
|
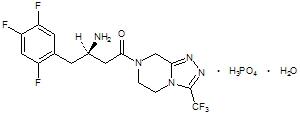
Sitagliptin phosphate monohydrate is a white to off-white, crystalline, non- hygroscopic powder. It is soluble in water and N,N-dimethyl formamide; slightly soluble in methanol; very slightly soluble in ethanol, acetone, and acetonitrile; and insoluble in isopropanol and isopropyl acetate.
Each film-coated tablet of JANUVIA contains 32.13, 64.25, or 128.5 mg of sitagliptin phosphate monohydrate, which is equivalent to 25, 50, or 100 mg, respectively, of free base and the following inactive ingredients: microcrystalline cellulose, anhydrous dibasic calcium phosphate, croscarmellose sodium, magnesium stearate, and sodium stearyl fumarate. In addition, the film coating contains the following inactive ingredients: polyvinyl alcohol, polyethylene glycol, talc, titanium dioxide, red iron oxide, and yellow iron oxide.
CLINICAL PHARMACOLOGY SECTION
12 CLINICAL PHARMACOLOGY
12.1 Mechanism of Action
Sitagliptin is a DPP-4 inhibitor, which is believed to exert its actions in patients with type 2 diabetes mellitus by slowing the inactivation of incretin hormones. Concentrations of the active intact hormones are increased by sitagliptin, thereby increasing and prolonging the action of these hormones. Incretin hormones, including glucagon-like peptide-1 (GLP-1) and glucose- dependent insulinotropic polypeptide (GIP), are released by the intestine throughout the day, and levels are increased in response to a meal. These hormones are rapidly inactivated by the enzyme, DPP-4. The incretins are part of an endogenous system involved in the physiologic regulation of glucose homeostasis. When blood glucose concentrations are normal or elevated, GLP-1 and GIP increase insulin synthesis and release from pancreatic beta cells by intracellular signaling pathways involving cyclic AMP. GLP-1 also lowers glucagon secretion from pancreatic alpha cells, leading to reduced hepatic glucose production. By increasing and prolonging active incretin levels, sitagliptin increases insulin release and decreases glucagon levels in the circulation in a glucose-dependent manner. Sitagliptin demonstrates selectivity for DPP-4 and does not inhibit DPP-8 or DPP-9 activity in vitro at concentrations approximating those from therapeutic doses.
12.2 Pharmacodynamics
General
In patients with type 2 diabetes mellitus, administration of sitagliptin led to inhibition of DPP-4 enzyme activity for a 24-hour period. After an oral glucose load or a meal, this DPP-4 inhibition resulted in a 2- to 3-fold increase in circulating levels of active GLP-1 and GIP, decreased glucagon concentrations, and increased responsiveness of insulin release to glucose, resulting in higher C-peptide and insulin concentrations. The rise in insulin with the decrease in glucagon was associated with lower fasting glucose concentrations and reduced glucose excursion following an oral glucose load or a meal.
In studies with healthy subjects, sitagliptin did not lower blood glucose or cause hypoglycemia.
Sitagliptin and Metformin hydrochloride Coadministration
In a two-day study in healthy subjects, sitagliptin alone increased active GLP-1 concentrations, whereas metformin alone increased active and total GLP-1 concentrations to similar extents. Coadministration of sitagliptin and metformin had an additive effect on active GLP-1 concentrations. Sitagliptin, but not metformin, increased active GIP concentrations. It is unclear how these findings relate to changes in glycemic control in patients with type 2 diabetes mellitus.
Cardiac Electrophysiology
In a randomized, placebo-controlled crossover study, 79 healthy subjects were administered a single oral dose of sitagliptin 100 mg, sitagliptin 800 mg (8 times the recommended dose), and placebo. At the recommended dose of 100 mg, there was no effect on the QTc interval obtained at the peak plasma concentration, or at any other time during the study. Following the 800 mg dose, the maximum increase in the placebo-corrected mean change in QTc from baseline was observed at 3 hours postdose and was 8.0 msec. This increase is not considered to be clinically significant. At the 800 mg dose, peak sitagliptin plasma concentrations were approximately 11 times higher than the peak concentrations following a 100-mg dose.
In patients with type 2 diabetes mellitus administered sitagliptin 100 mg (N=81) or sitagliptin 200 mg (N=63) daily, there were no meaningful changes in QTc interval based on ECG data obtained at the time of expected peak plasma concentration.
12.3 Pharmacokinetics
The pharmacokinetics of sitagliptin have been extensively characterized in healthy subjects and patients with type 2 diabetes mellitus. Following a single oral 100-mg dose to healthy volunteers, mean plasma AUC of sitagliptin was 8.52 μM•hr, Cmax was 950 nM, and apparent terminal half-life (t1/2) was 12.4 hours. Plasma AUC of sitagliptin increased in a dose-proportional manner and increased approximately 14% following 100 mg doses at steady-state compared to the first dose. The intra-subject and inter-subject coefficients of variation for sitagliptin AUC were small (5.8% and 15.1%). The pharmacokinetics of sitagliptin was generally similar in healthy subjects and in patients with type 2 diabetes mellitus.
Absorption
After oral administration of a 100 mg dose to healthy subjects, sitagliptin was rapidly absorbed with peak plasma concentrations (median Tmax) occurring 1 to 4 hours postdose. The absolute bioavailability of sitagliptin is approximately 87%.
Effect of Food
Coadministration of a high-fat meal with sitagliptin had no effect on the pharmacokinetics of sitagliptin.
Distribution
The mean volume of distribution at steady state following a single 100-mg intravenous dose of sitagliptin to healthy subjects is approximately 198 liters. The fraction of sitagliptin reversibly bound to plasma proteins is low (38%).
Elimination
Approximately 79% of sitagliptin is excreted unchanged in the urine with metabolism being a minor pathway of elimination. The apparent terminal t1/2 following a 100 mg oral dose of sitagliptin was approximately 12.4 hours and renal clearance was approximately 350 mL/min.
Metabolism
Following a [14C]sitagliptin oral dose, approximately 16% of the radioactivity was excreted as metabolites of sitagliptin. Six metabolites were detected at trace levels and are not expected to contribute to the plasma DPP-4 inhibitory activity of sitagliptin. In vitro studies indicated that the primary enzyme responsible for the limited metabolism of sitagliptin was CYP3A4, with contribution from CYP2C8.
Excretion
Following administration of an oral [14C]sitagliptin dose to healthy subjects, approximately 100% of the administered radioactivity was eliminated in feces (13%) or urine (87%) within one week of dosing.
Elimination of sitagliptin occurs primarily via renal excretion and involves active tubular secretion. Sitagliptin is a substrate for human organic anion transporter-3 (hOAT-3), which may be involved in the renal elimination of sitagliptin. The clinical relevance of hOAT-3 in sitagliptin transport has not been established. Sitagliptin is also a substrate of P-glycoprotein (P-gp), which may also be involved in mediating the renal elimination of sitagliptin. However, cyclosporine, a P-gp inhibitor, did not reduce the renal clearance of sitagliptin.
Specific Populations
Patients with Renal Impairment
An approximately 2-fold increase in the plasma AUC of sitagliptin was observed in patients with moderate renal impairment with eGFR of 30 to less than 45 mL/min/1.73 m2, and an approximately 4-fold increase was observed in patients with severe renal impairment, including patients with ESRD on hemodialysis, as compared to normal healthy control subjects.
Patients with Hepatic Impairment
In patients with moderate hepatic impairment (Child-Pugh score 7 to 9), mean AUC and Cmax of sitagliptin increased approximately 21% and 13%, respectively, compared to healthy matched controls following administration of a single 100-mg dose of sitagliptin. These differences are not considered to be clinically meaningful.
There is no clinical experience in patients with severe hepatic impairment (Child-Pugh score >9).
Effects of Age, Body Mass Index (BMI), Gender, and Race
Based on a population pharmacokinetic analysis or a composite analysis of available pharmacokinetic data, BMI, gender, and race do not have a clinically meaningful effect on the pharmacokinetics of sitagliptin. When the effects of age on renal function are taken into account, age alone did not have a clinically meaningful impact on the pharmacokinetics of sitagliptin based on a population pharmacokinetic analysis. Elderly subjects (65 to 80 years) had approximately 19% higher plasma concentrations of sitagliptin compared to younger subjects.
Drug Interaction Studies
In Vitro Assessment of Drug Interactions
Sitagliptin is not an inhibitor of CYP isozymes CYP3A4, 2C8, 2C9, 2D6, 1A2, 2C19 or 2B6, and is not an inducer of CYP3A4. Sitagliptin is a P-gp substrate, but does not inhibit P-gp mediated transport of digoxin. Based on these results, sitagliptin is considered unlikely to cause interactions with other drugs that utilize these pathways.
Sitagliptin is not extensively bound to plasma proteins. Therefore, the propensity of sitagliptin to be involved in clinically meaningful drug-drug interactions mediated by plasma protein binding displacement is very low.
In Vivo Assessment of Drug Interactions
Effects of Sitagliptin on Other Drugs
In clinical studies, sitagliptin did not meaningfully alter the pharmacokinetics of metformin, glyburide, simvastatin, rosiglitazone, digoxin, warfarin, or an oral contraceptive (ethinyl estradiol and norethindrone) (Table 4), providing in vivo evidence of a low propensity for causing drug interactions with substrates of CYP3A4, CYP2C8, CYP2C9, P-gp, and organic cationic transporter (OCT).
Table 4: Effect of Sitagliptin on Systemic Exposure of Coadministered Drugs|
Coadministered Drug |
Dose of Coadministered Drug* |
Dose of Sitagliptin* |
Geometric Mean Ratio | ||
|---|---|---|---|---|---|
|
AUC† |
C****max | ||||
| |||||
|
Digoxin |
0.25 mg‡ once daily for 10 days |
100 mg‡ once daily for 10 days |
Digoxin |
1.11§ |
1.18 |
|
Glyburide |
1.25 mg |
200 mg‡ once daily for 6 days |
Glyburide |
1.09 |
1.01 |
|
Simvastatin |
20 mg |
200 mg‡ once daily for 5 days |
Simvastatin |
0.85¶ |
0.80 |
|
Simvastatin Acid |
1.12¶ |
1.06 | |||
|
Rosiglitazone |
4 mg |
200 mg‡ once daily for 5 days |
Rosiglitazone |
0.98 |
0.99 |
|
Warfarin |
30 mg single dose on day 5 |
200 mg‡ once daily for 11 days |
S(-) Warfarin |
0.95 |
0.89 |
|
R(+) Warfarin |
0.99 |
0.89 | |||
|
Ethinyl estradiol and norethindrone |
21 days once daily of 35 µg ethinyl estradiol with norethindrone 0.5 mg × 7 days, 0.75 mg × 7 days, 1.0 mg × 7 days |
200 mg‡ once daily for 21 days |
Ethinyl estradiol |
0.99 |
0.97 |
|
Norethindrone |
1.03 |
0.98 | |||
|
Metformin HCl |
1000 mg‡ twice daily for 14 days |
50 mg‡ twice daily for 7 days |
Metformin |
1.02# |
0.97 |
Effects of Other Drugs on Sitagliptin
Clinical data described below suggest that sitagliptin is not susceptible to clinically meaningful interactions by coadministered medications (Table 5).
Table 5: Effect of Coadministered Drugs on Systemic Exposure of Sitagliptin|
Coadministered Drug |
Dose of Coadministered Drug* |
Dose of Sitagliptin* |
Geometric Mean Ratio | ||
|---|---|---|---|---|---|
|
AUC† |
C****max | ||||
| |||||
|
Cyclosporine |
600 mg once daily |
100 mg once daily |
Sitagliptin |
1.29 |
1.68 |
|
Metformin HCl |
1000 mg‡ twice daily for 14 days |
50 mg‡ twice daily for 7 days |
Sitagliptin |
1.02§ |
1.05 |
NONCLINICAL TOXICOLOGY SECTION
13 NONCLINICAL TOXICOLOGY
13.1 Carcinogenesis, Mutagenesis, Impairment of Fertility
A two-year carcinogenicity study was conducted in male and female rats given oral doses of sitagliptin of 50, 150, and 500 mg/kg/day. There was an increased incidence of combined liver adenoma/carcinoma in males and females and of liver carcinoma in females at 500 mg/kg. This dose results in exposures approximately 60 times the human exposure at the maximum recommended daily adult human dose (MRHD) of 100 mg/day based on AUC comparisons. Liver tumors were not observed at 150 mg/kg, approximately 20 times the human exposure at the MRHD. A two-year carcinogenicity study was conducted in male and female mice given oral doses of sitagliptin of 50, 125, 250, and 500 mg/kg/day. There was no increase in the incidence of tumors in any organ up to 500 mg/kg, approximately 70 times human exposure at the MRHD. Sitagliptin was not mutagenic or clastogenic with or without metabolic activation in the Ames bacterial mutagenicity assay, a Chinese hamster ovary (CHO) chromosome aberration assay, an in vitro cytogenetics assay in CHO, an in vitro rat hepatocyte DNA alkaline elution assay, and an in vivo micronucleus assay.
In rat fertility studies with oral gavage doses of 125, 250, and 1000 mg/kg, males were treated for 4 weeks prior to mating, during mating, up to scheduled termination (approximately 8 weeks total) and females were treated 2 weeks prior to mating through gestation day 7. No adverse effect on fertility was observed at 125 mg/kg (approximately 12 times human exposure at the MRHD of 100 mg/day based on AUC comparisons). At higher doses, nondose-related increased resorptions in females were observed (approximately 25 and 100 times human exposure at the MRHD based on AUC comparison).
HOW SUPPLIED SECTION
16 HOW SUPPLIED/STORAGE AND HANDLING
Tablets: JANUVIA, 100 mg, are beige, round, film-coated tablets with "277" on one side.
They are supplied as follows:
Bottles of approximately 1350 tablets, NDC 55154-5042-8
Storage
Store at 20-25°C (68-77°F), excursions permitted to 15-30°C (59-86°F). [See USP Controlled Room Temperature.]
INFORMATION FOR PATIENTS SECTION
17 PATIENT COUNSELING INFORMATION
Advise the patient to read the FDA-approved patient labeling (Medication Guide).
Pancreatitis
Inform patients that acute pancreatitis has been reported during postmarketing use of JANUVIA. Inform patients that persistent severe abdominal pain, sometimes radiating to the back, which may or may not be accompanied by vomiting, is the hallmark symptom of acute pancreatitis. Instruct patients to promptly discontinue JANUVIA and contact their physician if persistent severe abdominal pain occurs [see Warnings and Precautions (5.1)].
Heart Failure
Inform patients of the signs and symptoms of heart failure. Before initiating JANUVIA, ask patients about a history of heart failure or other risk factors for heart failure including moderate to severe renal impairment. Instruct patients to contact their health care provider as soon as possible if they experience symptoms of heart failure, including increasing shortness of breath, rapid increase in weight or swelling of the feet [see Warnings and Precautions (5.2)].
Hypoglycemia
Inform patients that the incidence of hypoglycemia is increased when JANUVIA is added to a sulfonylurea or insulin. Explain to patients receiving JANUVIA in combination with these medications the risks of hypoglycemia, its symptoms and treatment, and conditions that predispose to its development [see Warnings and Precautions (5.4)].
Hypersensitivity Reactions
Inform patients that allergic reactions have been reported during postmarketing use of JANUVIA. If symptoms of allergic reactions (including rash, hives, and swelling of the face, lips, tongue, and throat that may cause difficulty in breathing or swallowing) occur, patients must stop taking JANUVIA and seek medical advice promptly [see Warnings and Precautions (5.5)].
Severe and Disabling Arthralgia
Inform patients that severe and disabling joint pain may occur with this class of drugs. The time to onset of symptoms can range from one day to years. Instruct patients to seek medical advice if severe joint pain occurs [see Warnings and Precautions (5.6)].
Bullous Pemphigoid
Inform patients that bullous pemphigoid may occur with this class of drugs. Instruct patients to seek medical advice if blisters or erosions occur [see Warnings and Precautions (5.7)].
SPL MEDGUIDE SECTION
|
Medication Guide | ||||||
|---|---|---|---|---|---|---|
|
This Medication Guide has been approved by the U.S. Food and Drug Administration. | ||||||
|
Revised: 07/2022 | ||||||
|
Read this Medication Guide carefully before you start taking JANUVIA and each time you get a refill. There may be new information. This information does not take the place of talking with your doctor about your medical condition or your treatment. If you have any questions about JANUVIA, ask your doctor or pharmacist. | ||||||
|
What is the most important information I should know about JANUVIA? • Before you start taking JANUVIA, tell your doctor if you have ever had: | ||||||
|
• • |
• • |
• | ||||
|
Stop taking JANUVIA and call your doctor right away if you have pain in your stomach area (abdomen) that is severe and will not go away. The pain may be felt going from your abdomen through to your back. The pain may happen with or without vomiting. These may be symptoms of pancreatitis. | ||||||
|
• Before you start taking JANUVIA, tell your doctor if you have ever had heart failure or have problems with your kidneys. Contact your doctor right away if you have any of the following symptoms: | ||||||
|
• • • • | ||||||
|
What is JANUVIA? | ||||||
|
Who should not take JANUVIA? Symptoms of a serious allergic reaction to JANUVIA may include rash, raised red patches on your skin (hives), or swelling of the face, lips, tongue, and throat that may cause difficulty in breathing or swallowing. | ||||||
|
What should I tell my doctor before taking JANUVIA? • • • • • Tell your doctor about all the medicines you take, including prescription
and over-the-counter medicines, vitamins, and herbal supplements. | ||||||
|
How should I take JANUVIA? • • • • • • • • • • • | ||||||
|
What are the possible side effects of JANUVIA? • • • | ||||||
|
• • |
• • |
• • |
• • |
• • | ||
|
• • • The most common side effects of JANUVIA include upper respiratory infection,
stuffy or runny nose and sore throat, and headache. | ||||||
|
How should I store JANUVIA? | ||||||
|
General information about the safe and effective use of JANUVIA. | ||||||
|
What are the ingredients in JANUVIA? | ||||||
Rahway, NJ 07065, USA Cardinal Health Dublin, OH 43017 L7309 Rev. D For patent information:
www.msd.com/research/patent.
The trademarks depicted herein are owned by their respective companies.
Copyright © 2010-2022 Merck & Co., Inc., Rahway, NJ, USA, and its affiliates. |