
Tolvaptan
These highlights do not include all the information needed to use TOLVAPTAN TABLETS safely and effectively. See full prescribing information for TOLVAPTAN TABLETS. TOLVAPTAN tablets, for oral use Initial U.S. Approval: 2009
6f4c5679-8490-45ec-bd34-1a569e3af850
HUMAN PRESCRIPTION DRUG LABEL
Sep 5, 2022
Camber Pharmaceuticals, Inc.
DUNS: 826774775
Products 2
Detailed information about drug products covered under this FDA approval, including NDC codes, dosage forms, ingredients, and administration routes.
Tolvaptan
Product Details
FDA regulatory identification and product classification information
FDA Identifiers
Product Classification
Product Specifications
INGREDIENTS (10)
Tolvaptan
Product Details
FDA regulatory identification and product classification information
FDA Identifiers
Product Classification
Product Specifications
INGREDIENTS (9)
Drug Labeling Information
PACKAGE LABEL.PRINCIPAL DISPLAY PANEL
PACKAGE LABEL.PRINCIPAL DISPLAY PANEL
Tolvaptan Tablets 30 mg - carton label
Tolvaptan Tablets 15 mg - carton label
BOXED WARNING SECTION
WARNING: (A) INITIATE AND RE-INITIATE IN A HOSPITAL AND MONITOR SERUM
SODIUM (B) NOT FOR USE FOR AUTOSOMAL DOMINANT POLYCYSTIC KIDNEY DISEASE (ADPKD)
INDICATIONS & USAGE SECTION
1 INDICATIONS AND USAGE
Tolvaptan tablets are indicated for the treatment of clinically significant
hypervolemic and euvolemic hyponatremia (serum sodium <125 mEq/L or less
marked hyponatremia that is symptomatic and has resisted correction with fluid
restriction), including patients with heart failure and Syndrome of
Inappropriate Antidiuretic Hormone (SIADH).
Limitations of Use:
Patients requiring intervention to raise serum sodium urgently to prevent or
to treat serious neurological symptoms should not be treated with tolvaptan
tablets.
It has not been established that raising serum sodium with tolvaptan tablets
provide a symptomatic benefit to patients.
Tolvaptan tablets are a selective vasopressin V 2-receptor antagonist
indicated for the treatment of clinically significant hypervolemic and
euvolemic hyponatremia [serum sodium <125 mEq/L or less marked hyponatremia that is symptomatic and has resisted correction with fluid restriction],
including patients with heart failure and Syndrome of Inappropriate
Antidiuretic Hormone (SIADH) ( 1)
Limitations of Use:
- Patients requiring intervention to raise serum sodium urgently to prevent or to treat serious neurological symptoms should not be treated with tolvaptan tablets ( 1)
- It has not been established that tolvaptan tablets provide a symptomatic benefit to patients ( 1)
CONTRAINDICATIONS SECTION
4 CONTRAINDICATIONS
Tolvaptan tablets are contraindicated in the following conditions:
• Patients with autosomal dominant polycystic kidney disease (ADPKD) outside
of FDA-approved REMS [see Warnings and Precautions ( 5.2)]
• Unable to sense or respond to thirst
• Hypovolemic hyponatremia
• Taking strong CYP3A inhibitors [see Warnings and Precautions ( 5.5)]
• Anuria
• Hypersensitivity (e.g., anaphylactic shock, rash generalized) to tolvaptan
or any components of the product [see Adverse Reactions ( 6)]
• Use in patients with autosomal dominant polycystic kidney disease (ADPKD)
outside of FDA-approved REMS ( 4)
• Patients who are unable to respond appropriately to thirst ( 4)
• Hypovolemic hyponatremia ( 4)
• Concomitant use of strong CYP3A inhibitors ( 4)
• Anuria ( 4)
• Hypersensitivity ( 4)
WARNINGS AND PRECAUTIONS SECTION
5 WARNINGS AND PRECAUTIONS
5.1 Too Rapid Correction of Serum Sodium Can Cause Serious Neurologic
Sequelae
Osmotic demyelination syndrome is a risk associated with too rapid correction of hyponatremia (e.g., >12 mEq/L/24 hours). Osmotic demyelination results in dysarthria, mutism, dysphagia, lethargy, affective changes, spastic quadriparesis, seizures, coma or death. In susceptible patients, including those with severe malnutrition, alcoholism or advanced liver disease, slower rates of correction may be advisable. In controlled clinical trials in which tolvaptan was administered in titrated doses starting at 15 mg once daily, 7% of tolvaptan-treated subjects with a serum sodium <130 mEq/L had an increase in serum sodium greater than 8 mEq/L at approximately 8 hours and 2% had an increase greater than 12 mEq/L at 24 hours. Approximately 1% of placebo- treated subjects with a serum sodium <130 mEq/L had a rise greater than 8 mEq/L at 8 hours and no patient had a rise greater than 12 mEq/L/24 hours. Osmotic demyelination syndrome has been reported in association with tolvaptan tablets therapy [see Adverse Reactions ( 6.2)] .
Patients treated with tolvaptan tablets should be monitored to assess serum sodium concentrations and neurologic status, especially during initiation and after titration. Subjects with SIADH or very low baseline serum sodium concentrations may be at greater risk for too-rapid correction of serum sodium. In patients receiving tolvaptan tablets who develop too rapid a rise in serum sodium, discontinue or interrupt treatment with tolvaptan tablets and consider administration of hypotonic fluid. Fluid restriction during the first 24 hours of therapy with tolvaptan tablets may increase the likelihood of overly rapid correction of serum sodium and should generally be avoided. Co- administration of diuretics also increases the risk of too rapid correction of serum sodium and such patients should undergo close monitoring of serum sodium.
5.2 Liver Injury
Tolvaptan can cause serious and potentially fatal liver injury. In placebo-
controlled studies and an open-label extension study of chronically
administered tolvaptan in patients with ADPKD, cases of serious liver injury
attributed to tolvaptan, generally occurring during the first 18 months of
therapy, were observed. In postmarketing experience with tolvaptan in ADPKD,
acute injury resulting in liver failure requiring liver transplantation has
been reported. Tolvaptan should not be used to treat ADPKD outside of the FDA-
approved risk evaluation and mitigation strategy (REMS) for ADPKD patients
[see Contraindications ( 4)].
Patients with symptoms that may indicate liver injury, including fatigue,
anorexia, right upper abdominal discomfort, dark urine or jaundice should
discontinue treatment with tolvaptan tablets.
Limit duration of therapy with tolvaptan tablets to 30 days. Avoid use in
patients with underlying liver disease, including cirrhosis, because the
ability to recover from liver injury may be impaired [see Adverse Reactions ( 6.1)].
5.3 Dehydration and Hypovolemia
Tolvaptan tablets therapy induces copious aquaresis, which is normally partially offset by fluid intake. Dehydration and hypovolemia can occur, especially in potentially volume-depleted patients receiving diuretics or those who are fluid restricted. In multiple-dose, placebo-controlled trials in which 607 hyponatremic patients were treated with tolvaptan, the incidence of dehydration was 3.3% for tolvaptan and 1.5% for placebo-treated patients. In patients receiving tolvaptan tablets who develop medically significant signs or symptoms of hypovolemia, interrupt or discontinue tolvaptan tablets therapy and provide supportive care with careful management of vital signs, fluid balance and electrolytes. Fluid restriction during therapy with tolvaptan tablets may increase the risk of dehydration and hypovolemia. Patients receiving tolvaptan tablets should continue ingestion of fluid in response to thirst.
5.4 Co-administration with Hypertonic Saline
Concomitant use with hypertonic saline is not recommended.
5.5 Drug Interactions
Tolvaptan is a substrate of CYP3A. Moderate to strong CYP3A inhibitors can lead to a marked increase in tolvaptan concentrations [see Drug Interactions ( 7.1)]. Do not use tolvaptan tablets with strong inhibitors of CYP3A [see Contraindications ( 4)] and avoid concomitant use with moderate CYP3A inhibitors.
5.6 Hyperkalemia or Drugs that Increase Serum Potassium
Treatment with tolvaptan is associated with an acute reduction of the extracellular fluid volume which could result in increased serum potassium. Serum potassium levels should be monitored after initiation of tolvaptan treatment in patients with a serum potassium >5 mEq/L as well as those who are receiving drugs known to increase serum potassium levels.
5.7 Acute Urinary Retention with Outflow Obstruction
Patients with partial obstruction of urinary outflow, for example, patients with prostatic hypertrophy or impairment of micturition, have an increased risk of developing acute retention. Do not administer tolvaptan in patients with uncorrected urinary outflow obstruction.
• Liver injury: Limit treatment duration to 30 days. If hepatic injury is
suspected, discontinue tolvaptan tablets. Avoid use in patients with
underlying liver disease ( 5.2)
• Dehydration and hypovolemia may require intervention ( 5.3)
• Avoid use with hypertonic saline ( 5.4)
• Avoid use with moderate to strong CYP3A inhibitors ( 5.5)
• Monitor serum potassium in patients with potassium >5 mEq/L or on drugs
known to increase potassium ( 5.6)
• Urinary outflow obstruction: Urinary output must be secured ( 5.7)
ADVERSE REACTIONS SECTION
6 ADVERSE REACTIONS
6.1 Clinical Trials Experience
Because clinical trials are conducted under widely varying conditions, adverse
reactions rates observed in the clinical trials of a drug cannot be directly
compared to rates in the clinical trials of another drug and may not reflect
the rates observed in practice. The adverse event information from clinical
trials does, however, provide a basis for identifying the adverse events that
appear to be related to drug use and for approximating rates.
In multiple-dose, placebo-controlled trials, 607 hyponatremic patients (serum
sodium <135 mEq/L) were treated with tolvaptan tablets. The mean age of these
patients was 62 years; 70% of patients were male and 82% were Caucasian. One
hundred eighty-nine (189) tolvaptan-treated patients had a serum sodium <130
mEq/L, and 52 patients had a serum sodium <125 mEq/L. Hyponatremia was
attributed to cirrhosis in 17% of patients, heart failure in 68% and
SIADH/other in 16%. Of these patients, 223 were treated with the recommended
dose titration (15 mg titrated to 60 mg as needed to raise serum sodium).
Overall, over 4,000 patients have been treated with oral doses of tolvaptan in
open-label or placebo-controlled clinical trials. Approximately 650 of these
patients had hyponatremia; approximately 219 of these hyponatremic patients
were treated with tolvaptan for 6 months or more.
The most common adverse reactions (incidence ≥5% more than placebo) seen in
two 30-day, double-blind, placebo-controlled hyponatremia trials in which
tolvaptan was administered in titrated doses (15 mg to 60 mg once daily) were
thirst, dry mouth, asthenia, constipation, pollakiuria or polyuria and
hyperglycemia. In these trials, 10% (23/223) of tolvaptan-treated patients
discontinued treatment because of an adverse event, compared to 12% (26/220)
of placebo-treated patients; no adverse reaction resulting in discontinuation
of trial medication occurred at an incidence of >1% in tolvaptan-treated
patients.
Table 1 lists the adverse reactions reported in tolvaptan-treated patients
with hyponatremia (serum sodium <135 mEq/L) and at a rate at least 2% greater
than placebo-treated patients in two 30-day, double-blind, placebo-controlled
trials. In these studies, 223 patients were exposed to tolvaptan (starting
dose 15 mg, titrated to 30 and 60 mg as needed to raise serum sodium). Adverse
events resulting in death in these trials were 6% in tolvaptan-treated
patients and 6% in placebo-treated patients.
Table 1. Adverse Reactions (>2% more than placebo) in Tolvaptan-Treated
Patients in Double-Blind, Placebo-Controlled Hyponatremia Trials
|
System Organ Class |
** Tolvaptan** |
Placebo |
|
** Gastrointestinal Disorders** | ||
|
Dry mouth |
28 (13) |
9 (4) |
|
Constipation |
16 (7) |
4 (2) |
|
** General Disorders and Administration Site Conditions** | ||
|
Thirst * |
35 (16) |
11 (5) |
|
Asthenia |
19 (9) |
9 (4) |
|
Pyrexia |
9 (4) |
2 (1) |
|
Metabolism and Nutrition Disorders | ||
|
Hyperglycemia † |
14 (6) |
2 (1) |
|
Anorexia ‡ |
8 (4) |
2 (1) |
|
** Renal and Urinary Disorders** | ||
|
Pollakiuria or polyuria § |
25 (11) |
7 (3) |
The following terms are subsumed under the referenced ADR in Table 1:
*polydipsia; †diabetes mellitus; ‡decreased appetite; §urine output increased, micturition urgency, nocturia
In a subgroup of patients with hyponatremia (N = 475, serum sodium <135 mEq/L) enrolled in a double-blind, placebo-controlled trial (mean duration of treatment was 9 months) of patients with worsening heart failure, the following adverse reactions occurred in tolvaptan-treated patients at a rate at least 2% greater than placebo: mortality (42% tolvaptan, 38% placebo), nausea (21% tolvaptan, 16% placebo), thirst (12% tolvaptan, 2% placebo), dry mouth (7% tolvaptan, 2% placebo) and polyuria or pollakiuria (4% tolvaptan, 1% placebo).
Gastrointestinal bleeding in patients with cirrhosis
In patients with cirrhosis treated with tolvaptan in the hyponatremia trials,
gastrointestinal bleeding was reported in 6 out of 63 (10%) tolvaptan-treated
patients and 1 out of 57 (2%) placebo treated patients.
The following adverse reactions occurred in <2% of hyponatremic patients
treated with tolvaptan tablets and at a rate greater than placebo in double-
blind placebo-controlled trials (N = 607 tolvaptan; N = 518 placebo) or in <2%
of patients in an uncontrolled trial of patients with hyponatremia (N = 111)
and are not mentioned elsewhere in the label.
Blood and Lymphatic System Disorders: Disseminated intravascular coagulation
Cardiac Disorders: Intracardiac thrombus, ventricular fibrillation
Investigations: Prothrombin time prolonged
Gastrointestinal Disorders: Ischemic colitis
Metabolism and Nutrition Disorders: Diabetic ketoacidosis
Musculoskeletal and Connective Tissue Disorders: Rhabdomyolysis
Nervous System: Cerebrovascular accident
Renal and Urinary Disorders: Urethral hemorrhage
Reproductive System and Breast Disorders (female): Vaginal hemorrhage
Respiratory, Thoracic, and Mediastinal Disorders: Pulmonary embolism,
respiratory failure
Vascular disorder: Deep vein thrombosis
6.2 Postmarketing Experience
The following adverse reactions have been identified during post-approval use of tolvaptan tablets. Because these reactions are reported voluntarily from a population of an unknown size, it is not always possible to reliably estimate their frequency or establish a causal relationship to drug exposure.
Neurologic: Osmotic demyelination syndrome
Investigations: Hypernatremia
Removal of excess free body water increases serum osmolality and serum sodium concentrations. All patients treated with tolvaptan, especially those whose serum sodium levels become normal, should continue to be monitored to ensure serum sodium remains within normal limits. If hypernatremia is observed, management may include dose decreases or interruption of tolvaptan treatment, combined with modification of free-water intake or infusion. During clinical trials of hyponatremic patients, hypernatremia was reported as an adverse event in 0.7% of patients receiving tolvaptan vs. 0.6% of patients receiving placebo; analysis of laboratory values demonstrated an incidence of hypernatremia of 1.7% in patients receiving tolvaptan vs. 0.8% in patients receiving placebo.
Immune System Disorders: Hypersensitivity reactions including anaphylactic shock and rash generalized.
Most common adverse reactions (≥5% placebo) are thirst, dry mouth, asthenia,
constipation, pollakiuria or polyuria, and hyperglycemia ( 6.1)
To report SUSPECTED ADVERSE REACTIONS, contact Annora Pharma Private Limited
at 1-866-495-1995 or FDA at 1-800-FDA-1088 (www.fda.gov/medwatch).
DRUG INTERACTIONS SECTION
7 DRUG INTERACTIONS
7.1 CYP3A Inhibitors and Inducers
Strong CYP3A Inhibitors
Tolvaptan's AUC was 5.4 times as large and C max was 3.5 times as large after
co-administration of tolvaptan and 200 mg ketoconazole [see Warnings and Precautions ( 5.5) and Clinical Pharmacology ( 12.3)] . Larger doses of the
strong CYP3A inhibitor would be expected to produce larger increases in
tolvaptan exposure. Concomitant use of tolvaptan with strong CYP3A inhibitors
is contraindicated [see Contraindications ( 4)] .
Moderate CYP3A Inhibitors
A substantial increase in the exposure to tolvaptan would be expected when
tolvaptan is co-administered with moderate CYP3A inhibitors. Avoid co-
administration of tolvaptan with moderate CYP3A inhibitors [see Warnings and Precautions ( 5.5)].
Patients should avoid grapefruit juice beverages while taking tolvaptan [see Clinical Pharmacology ( 12.3)] .
Strong CYP3A Inducers
Co-administration of tolvaptan with strong CYP3A inducers reduces exposure to
tolvaptan [see Clinical Pharmacology ( 12.3)] . Avoid concomitant use of
tolvaptan with strong CYP3A inducers.
7.2 Angiotensin Receptor Blockers, Angiotensin Converting Enzyme Inhibitors
and Potassium Sparing Diuretics
Although specific interaction studies were not performed, in clinical studies, tolvaptan was used concomitantly with beta-blockers, angiotensin receptor blockers, angiotensin converting enzyme inhibitors and potassium sparing diuretics. Adverse reactions of hyperkalemia were approximately 1 to 2% higher when tolvaptan was administered with angiotensin receptor blockers, angiotensin converting enzyme inhibitors and potassium sparing diuretics compared to administration of these medications with placebo. Serum potassium levels should be monitored during concomitant drug therapy.
7.3 V 2-Receptor Agonist
As a V 2-receptor antagonist, tolvaptan may interfere with the V 2-agonist activity of desmopressin (dDAVP). Avoid concomitant use of tolvaptan with a V 2-agonist.
Avoid concomitant use with:
• Moderate CYP3A inhibitors ( 7.1)
• Strong CYP3A inducers ( 7.1)
• V 2-receptor antagonists ( 7.3)
Monitor serum potassium during concomitant therapy with ( 7.2):
• Angiotensin receptor blockers
• Angiotensin converting enzyme inhibitors
• Potassium sparing diuretics
RECENT MAJOR CHANGES SECTION
RECENT MAJOR CHANGES
Warnings and Precautions ( 5.5, 5.7) 04/2021
DOSAGE & ADMINISTRATION SECTION
2 DOSAGE AND ADMINISTRATION
2.1 Recommended Dosage
Patients should be in a hospital for initiation and re-initiation of therapy
to evaluate the therapeutic response and because too rapid correction of
hyponatremia can cause osmotic demyelination resulting in dysarthria, mutism,
dysphagia, lethargy, affective changes, spastic quadriparesis, seizures, coma
and death.
The usual starting dose for tolvaptan tablets is 15 mg administered once daily
without regard to meals. Increase the dose to 30 mg once daily, after at least
24 hours, to a maximum of 60 mg once daily, as needed to achieve the desired
level of serum sodium. Do not administer tolvaptan tablets for more than 30
days to minimize the risk of liver injury [see Warnings and Precautions ( 5.2)].
During initiation and titration, frequently monitor for changes in serum
electrolytes and volume. Avoid fluid restriction during the first 24 hours of
therapy. Patients receiving tolvaptan tablets should be advised that they can
continue ingestion of fluid in response to thirst [see Warnings and Precautions ( 5.1)].
2.2 Drug Withdrawal
Following discontinuation from tolvaptan tablets, patients should be advised to resume fluid restriction and should be monitored for changes in serum sodium and volume status.
- Tolvaptan tablets should be initiated and re-initiated in a hospital ( 2.1)
- The recommended starting dose is 15 mg once daily. Dosage may be increased at intervals ≥24 hr to 30 mg once daily, and to a maximum of 60 mg once daily as needed to raise serum sodium. ( 2.1)
DOSAGE FORMS & STRENGTHS SECTION
3 DOSAGE FORMS AND STRENGTHS
Tolvaptan tablets are available in the following dosage forms and strengths:
- 15 mg tablets are white to off white colored, triangular, bevel edged, biconvex tablets debossed with 'H' on one side and 'T9' on the other side.
- 30 mg tablets are blue, round, bevel edged, biconvex tablets debossed with 'H' on one side and 'T10' on the other side.
- Tablets: 15 mg and 30 mg ( 3)
USE IN SPECIFIC POPULATIONS SECTION
8 USE IN SPECIFIC POPULATIONS
8.1 Pregnancy
Risk Summary
Available data with tolvaptan use in pregnant women are insufficient to
determine if there is a drug-associated risk of adverse developmental
outcomes. Tolvaptan did not cause any developmental toxicity in rats or in
rabbits at exposures approximately 2.8 and 0.8 times, respectively, the
exposure in congestive heart failure (CHF) patients at the maximum recommended
human dose (MRHD) of 60 mg once daily. However, effects on embryo-fetal
development occurred in both species at doses causing significant maternally
toxic doses. In rats, reduced fetal weights and delayed fetal ossification
occurred at 11 times the exposure in CHF patients, based on AUC. In rabbits,
increased abortions, embryo-fetal death, fetal microphthalmia, open eyelids,
cleft palate, brachymelia and skeletal malformations occurred at approximately
1.6 times the exposure in CHF patients (see Data).
The estimated background risk of major birth defects and miscarriage for the
indicated population is unknown. All pregnancies have a background risk of
birth defect, loss, or other adverse outcomes. The estimated background risk
of major birth defects and miscarriage in the U.S. general population is 2 to
4% and 15 to 20% of clinically recognized pregnancies, respectively.
Data
Animal Data
Oral administration of tolvaptan during the period of organogenesis in
Sprague-Dawley rats produced no evidence of teratogenesis at doses up to 100
mg/kg/day. Delayed ossification was seen at 1000 mg/kg, which is approximately
11 times the exposure in CHF patients at the MRHD of 60 mg (AUC 24h 10271
ng*h/mL). The fetal effects are likely secondary to maternal toxicity
(decreased food intake and low body weights). In a prenatal and postnatal
study in rats, tolvaptan had no effect on physical development, reflex
function, learning ability or reproductive performance at doses up to 1000
mg/kg/day (11 times the exposure in CHF patients at the MRHD of 60 mg).
In rabbits, teratogenicity (microphthalmia, embryo-fetal mortality, cleft
palate, brachymelia and skeletal malformations) was observed in rabbits at
1000 mg/kg (approximately 1.6 times the exposure in CHF patients at the MRHD
of 60 mg dose). This dose also caused maternal toxicity (lower body weight
gains and food consumption).
8.2 Lactation
Risk Summary
There are no data on the presence of tolvaptan or its metabolites in human
milk, the effects on the breastfed infant, or the effects on milk production.
Tolvaptan is present in rat milk ( see Data). When a drug is present in animal
milk, it is possible that the drug will be present in human milk, but relative
levels may vary ( see Data). Because of the potential for serious adverse
reactions, including electrolyte abnormalities (e.g., hypernatremia),
hypotension, and volume depletion in breastfed infants, advise women not to
breastfeed during treatment with tolvaptan.
Data
In lactating rats administered radiolabeled tolvaptan, lacteal radioactivity
concentrations reached the highest level at 8 hours after administration and
then decreased gradually with time with a half-life of 27.3 hours. The level
of activity in milk ranged from 1.5- to 15.8-fold those in maternal blood over
a period of 72 hours post-dose. Increased perinatal death and decreased body
weight of the offspring were observed during the lactation period and after
weaning at approximately 11 times the exposure in CHF patients at the MRHD of
60 mg.
8.4 Pediatric Use
Safety and effectiveness of tolvaptan tablets in pediatric patients have not been established.
8.5 Geriatric Use
Of the total number of hyponatremic subjects treated with tolvaptan tablets in clinical studies, 42% were 65 years old and over, while 19% were 75 years old and over. No overall differences in safety or effectiveness were observed between these subjects and younger subjects, and other reported clinical experience has not identified differences in responses between the elderly and younger patients, but greater sensitivity of some older individuals cannot be ruled out. Increasing age has no effect on tolvaptan plasma concentrations.
8.6 Use in Patients with Hepatic Impairment
Moderate and severe hepatic impairment do not affect exposure to tolvaptan to a clinically relevant extent. Avoid use of tolvaptan in patients with underlying liver disease.
8.7 Use in Patients with Renal Impairment
No dose adjustment is necessary based on renal function. There are no clinical trial data in patients with CrCl <10 mL/min, and, because drug effects on serum sodium levels are likely lost at very low levels of renal function, use in patients with a CrCl <10 mL/min is not recommended. No benefit can be expected in patients who are anuric [see Contraindications ( 4) and Clinical Pharmacology ( 12.3)] .
• Pregnancy: May cause fetal harm ( 8.1)
• Lactation: Breastfeeding not recommended ( 8.2)
• Pediatric Use: There are no studies ( 8.4)
OVERDOSAGE SECTION
10 OVERDOSAGE
Single oral doses up to 480 mg (8 times the maximum recommended daily dose)
and multiple doses up to 300 mg once daily for 5 days have been well tolerated
in studies in healthy subjects. There is no specific antidote for tolvaptan
intoxication. The signs and symptoms of an acute overdose can be anticipated
to be those of excessive pharmacologic effect: a rise in serum sodium
concentration, polyuria, thirst, and dehydration/hypovolemia.
No mortality was observed in rats or dogs following single oral doses of 2000
mg/kg (maximum feasible dose). A single oral dose of 2000 mg/kg was lethal in
mice, and symptoms of toxicity in affected mice included decreased locomotor
activity, staggering gait, tremor and hypothermia.
In patients with suspected tolvaptan overdosage, assessment of vital signs,
electrolyte concentrations, ECG and fluid status are recommended. Continue
replacement of water and electrolytes until aquaresis abates.
Dialysis may not be effective in removing tolvaptan because of its high
binding affinity for human plasma protein (>98%).
DESCRIPTION SECTION
11 DESCRIPTION
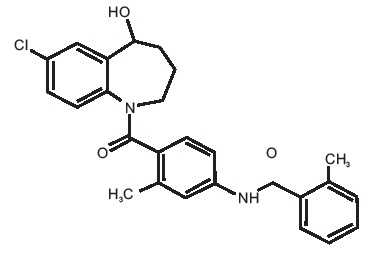
Tolvaptan, a selective vasopressin V 2-receptor antagonist in tablets for oral use available in 15 mg or 30 mg strength. Tolvaptan is (±) 4’-[(7-Chloro-2,3,4,5-tetrahydro-5-hydroxy-1H-benzazepin-1-yl) carbonyl]-O-tolu-m-toluidide. The empirical formula is C 26H 25ClN 2O 3. Molecular weight is 448.94. The chemical structure is:
Inactive ingredients include corn starch, croscarmellose sodium, lactose monohydrate, magnesium stearate, methyl alcohol, methylene chloride, microcrystalline cellulose and povidone. 30 mg contains FD&C Blue No # 2/Indigo caramine Aluminum Lake as colorant.
CLINICAL PHARMACOLOGY SECTION
12 CLINICAL PHARMACOLOGY
12.1 Mechanism of Action
Tolvaptan is a selective vasopressin V 2-receptor antagonist with an affinity for the V 2-receptor that is 1.8 times that of native arginine vasopressin (AVP). Tolvaptan affinity for the V 2-receptor is 29 times greater than for the V 1a-receptor. When taken orally, 15 to 60 mg doses of tolvaptan antagonize the effect of vasopressin and cause an increase in urine water excretion that results in an increase in free water clearance (aquaresis), a decrease in urine osmolality, and a resulting increase in serum sodium concentrations. Urinary excretion of sodium and potassium and plasma potassium concentrations are not significantly changed. Tolvaptan metabolites have no or weak antagonist activity for human V 2-receptors compared with tolvaptan.
12.2 Pharmacodynamics
In healthy subjects receiving a single dose of tolvaptan tablets 60 mg, the
onset of the aquaretic and sodium increasing effects occurs within 2 to 4
hours post-dose. A peak effect of about a 6 mEq increase in serum sodium and
about 9 mL/min increase in urine excretion rate is observed between 4 and 8
hours post-dose; thus, the pharmacological activity lags behind the plasma
concentrations of tolvaptan. About 60% of the peak effect on serum sodium is
sustained at 24 hours post-dose, but the urinary excretion rate is no longer
elevated by this time. Doses above 60 mg tolvaptan do not increase aquaresis
or serum sodium further. The effects of tolvaptan in the recommended dose
range of 15 to 60 mg once daily appear to be limited to aquaresis and the
resulting increase in sodium concentration.
Plasma concentrations of native AVP may increase (avg. 2 to 9 pg/mL) with
tolvaptan administration.
Cardiac Electrophysiology
No prolongation of the QT interval was observed with tolvaptan following
multiple doses of 300 mg/day for 5 days.
12.3 Pharmacokinetics
In healthy subjects, the pharmacokinetics of tolvaptan after single doses of
up to 480 mg and multiple doses up to 300 mg once daily have been examined. In
hyponatremia subjects, single and multiple doses up to 60 mg have been
studied.
Absorption
In healthy subjects, peak concentrations of tolvaptan are observed between 2
and 4 hours post-dose. Peak concentrations increase less than dose
proportionally with doses greater than 240 mg.
The absolute bioavailability of tolvaptan decreases with increasing doses. The
absolute bioavailability of tolvaptan following an oral dose of 30 mg is 56%
(range 42 to 80%).
Co-administration of 90 mg tolvaptan with a high-fat meal (~1000 calories, of
which 50% are from fat) doubles peak concentrations but has no effect on the
AUC of tolvaptan; tolvaptan may be administered with or without food.
Distribution
Tolvaptan binds to both albumin and α1-acid glycoprotein and the overall
protein binding is >98%; binding is not affected by disease state. The volume
of distribution of tolvaptan is about 3 L/kg. The pharmacokinetic properties
of tolvaptan are stereospecific, with a steady-state ratio of the S-(-) to the
R-(+) enantiomer of about 3. When administered as multiple once-daily 300 mg
doses to healthy subjects or to patients with congestive heart failure or
ADPKD, tolvaptan’s accumulation factor is <1.2. There is marked inter-subject
variation in peak and average exposure to tolvaptan with a percent coefficient
of variation ranging between 30 and 60%.
Metabolism and Elimination
Tolvaptan is metabolized almost exclusively by CYP3A. Fourteen metabolites
have been identified in plasma, urine and feces; all but one were also
metabolized by CYP3A and none are pharmacodynamically active. After oral
administration of radiolabeled tolvaptan, tolvaptan was a minor component in
plasma, representing 3% of total plasma radioactivity; the oxobutyric acid
metabolite was present at 52.5% of total plasma radioactivity with all other
metabolites present at lower concentrations than tolvaptan. The oxobutyric
acid metabolite shows a plasma half-life of ~180 h. About 40% of radioactivity
was recovered in urine (<1% as unchanged tolvaptan) and 59% in feces (19% as
unchanged tolvaptan). Following intravenous infusion, tolvaptan half-life is
approximately 3 hours. Following single oral doses to healthy subjects, the
estimated half-life of tolvaptan increases from 3 hours for a 15 mg dose to
approximately 12 hours for 120 mg and higher doses due to more prolonged
absorption of tolvaptan at higher doses; apparent clearance is approximately 4
mL/min/kg and does not appear to change with increasing dose.
Specific Populations
Hyponatremia
In patients with hyponatremia of any origin the clearance of tolvaptan is
reduced to about 2 mL/min/kg.
Hepatic Impairment
Moderate or severe hepatic impairment or congestive heart failure decrease the
clearance and increase the volume of distribution of tolvaptan, but the
respective changes are not clinically relevant. Exposure and response to
tolvaptan in subjects with creatinine clearance ranging between 79 and 10
mL/min and patients with normal renal function are not different.
Renal Impairment
In a study in patients with creatinine clearances ranging from 10 to 124
mL/min administered a single dose of 60 mg tolvaptan, AUC and C max of plasma
tolvaptan were less than doubled in patients with severe renal impairment
(creatinine clearance <30 mL/min) relative to the controls. The peak increase
in serum sodium was 5 to 6 mEq/L, regardless of renal function, but the onset
and offset of tolvaptan's effect on serum sodium were slower in patients with
severe renal impairment [see Use in Specific Populations ( 8.7)].
Drug Interaction Studies
Impact of Other Drugs on Tolvaptan
Strong CYP3A Inhibitors
Ketoconazole: Tolvaptan’s C max and AUC were, respectively, 3.5 times and 5.4
times as high following ketoconazole 200 mg given one day prior to and
concomitantly with 30 mg tolvaptan [see Contraindications ( 4), Warnings and Precautions ( 5.5) and Drug Interactions ( 7.1)] .
Moderate CYP3A4 inhibitors
Fluconazole: Fluconazole 400 mg given one day prior and 200 mg given
concomitantly produced an 80% and 200% increase in tolvaptan C max and AUC,
respectively.
Grapefruit Juice: Co-administration of grapefruit juice and tolvaptan results
in an increase in C max and AUC of 90% and 60% for tolvaptan, respectively
[see Drug Interactions ( 7.1)] .
CYP3A4 Inducers
Rifampin: Rifampin 600 mg once daily for 7 days followed by a single 240 mg
dose of tolvaptan decreased both tolvaptan C max and AUC about 85%.
Other Drugs
Co-administration of lovastatin, digoxin, furosemide, and hydrochlorothiazide
with tolvaptan has no clinically relevant impact on the exposure to tolvaptan.
Impact of Tolvaptan on Other Drugs
CYP3A Substrates
Tolvaptan is a weak inhibitor of CYP3A. Co-administration of lovastatin and
tolvaptan increases the exposure to lovastatin and its active metabolite
lovastatin-β hydroxyacid by factors of 1.4 and 1.3, respectively. This is not
a clinically relevant change.
P-gp Substrates
Digoxin: Digoxin 0.25 mg was administered once daily for 12 days. Tolvaptan 60
mg, was co-administered once daily on Days 8 to 12. Digoxin C max and AUC were
increased 30% and 20%, respectively.
Transporter Substrates
Tolvaptan is a substrate of P-gp and an inhibitor of P-gp and BCRP. The
oxobutyric acid metabolite of tolvaptan is an inhibitor of OATP1B1 and OAT3.
Co-administration of tolvaptan with rosuvastatin (BCRP substrate) did not have
a clinically significant effect on rosuvastatin exposure. Rosuvastatin C max
and AUC t increased 54% and 69%, respectively. Administration of rosuvastatin
(OATP1B1 substrate) or furosemide (OAT3 substrate) to healthy subjects with
elevated oxobutyric acid metabolite plasma concentrations did not meaningfully
alter the pharmacokinetics of rosuvastatin or furosemide.
Other Drugs
Co-administration of tolvaptan does not appear to alter the pharmacokinetics
of warfarin, furosemide, hydrochlorothiazide, or amiodarone (or its active
metabolite, desethylamiodarone) to a clinically significant degree.
NONCLINICAL TOXICOLOGY SECTION
13 NONCLINICAL TOXICOLOGY
13.1 Carcinogenesis, Mutagenesis, Impairment of Fertility
Carcinogenesis
The carcinogenic potential of tolvaptan was assessed in 2-year carcinogenicity
studies in mice and rats. Tolvaptan did not increase tumors in male or female
rats at doses up to 1000 mg/kg/day (1.24 to 3.26 times the exposure in CHF
patients based on AUC at the MRHD of 60 mg), in male mice at doses up to 60
mg/kg/day (0.3 times the exposure in CHF patients at the MRHD) and to female
mice at doses up to 100 mg/kg/day (0.4 times the exposure in CHF patients at
the MRHD).
Mutagenesis
Tolvaptan was not clastogenic in the in vitro chromosomal aberration test in
Chinese hamster lung fibroblast cells or the in vivo rat micronucleus assay
and was not mutagenic in the in vitro bacterial reverse mutation assays.
Impairment of fertility
In a fertility study in which male and female rats were administered tolvaptan
orally at 100, 300 or 1000 mg/kg/day, altered estrous cycles due to
prolongation of diestrus were observed in dams given 300 and 1000 mg/kg/day
(6.2 and 11 times the exposure in CHF patients at the 60 mg dose). Tolvaptan
had no effect on copulation or fertility indices. There were also no effects
on the incidences of early or late resorption, dead fetuses, pre- or post-
implantation loss, external anomalies, or fetal body weights.
CLINICAL STUDIES SECTION
14 CLINICAL STUDIES
14.1 Hyponatremia
In two double-blind, placebo-controlled, multi-center studies (SALT-1 and
SALT-2), a total of 424 patients with euvolemic or hypervolemic hyponatremia
(serum sodium <135 mEq/L) resulting from a variety of underlying causes (heart
failure, liver cirrhosis, syndrome of inappropriate antidiuretic hormone
[SIADH] and others) were treated for 30 days with tolvaptan or placebo, then
followed for an additional 7 days after withdrawal. Symptomatic patients,
patients likely to require saline therapy during the course of therapy,
patients with acute and transient hyponatremia associated with head trauma or
postoperative state and patients with hyponatremia due to primary polydipsia,
uncontrolled adrenal insufficiency or uncontrolled hypothyroidism were
excluded. Patients were randomized to receive either placebo (N = 220) or
tolvaptan (N = 223) at an initial oral dose of 15 mg once daily. The mean
serum sodium concentration at study entry was 129 mEq/L. Fluid restriction was
to be avoided if possible during the first 24 hours of therapy to avoid overly
rapid correction of serum sodium, and during the first 24 hours of therapy 87%
of patients had no fluid restriction. Thereafter, patients could resume or
initiate fluid restriction (defined as daily fluid intake of ≤1.0 liter/day)
as clinically indicated.
The dose of tolvaptan could be increased at 24-hour intervals to 30 mg once
daily, then to 60 mg once daily, until either the maximum dose of 60 mg or
normonatremia (serum sodium >135 mEq/L) was reached. Serum sodium
concentrations were determined at 8 hours after study drug initiation and
daily up to 72 hours, within which time titration was typically completed.
Treatment was maintained for 30 days with additional serum sodium assessments
on Days 11, 18, 25 and 30. On the day of study discontinuation, all patients
resumed previous therapies for hyponatremia and were reevaluated 7 days later.
The primary endpoint for these studies was the average daily AUC for change in
serum sodium from baseline to Day 4 and baseline to Day 30 in patients with a
serum sodium less than 135 mEq/L. Compared to placebo, tolvaptan caused a
statistically greater increase in serum sodium ( p <0.0001) during both
periods in both studies (see Table 2). For patients with a serum sodium of
<130 mEq/L or <125 mEq/L, the effects at Day 4 and Day 30 remained significant
(see Table 2). This effect was also seen across all disease etiology subsets
(e.g., CHF, cirrhosis, SIADH/other).
Table 2. Effects of Treatment with Tolvaptan 15 mg/day to 60 mg/day
|
Tolvaptan 15 mg/day to 60 mg/day |
Placebo |
Estimated Effect (95% CI) | |
|
Subjects with Serum Sodium <135 mEq/L (ITT population) | |||
|
Change in average daily serum [Na+] Mean (SD) N |
4.0 (2.8) 213 |
0.4 (2.4) 203 |
3.7 (3.3 to 4.2) p <0.0001 |
|
Change in average daily serum [Na+] Mean (SD) N |
6.2 (4.0) 213 |
1.8 (3.7) 203 |
4.6 (3.9 to 5.2) p <0.0001 |
|
Percent of Patients Needing Fluid Restriction* |
14% 30/215 |
25% 51/206 |
p=0.0017 |
|
Subgroup with Serum Sodium <130 mEq/L | |||
|
Change in average daily serum [Na+] Mean (SD) N |
4.8 (3.0) 110 |
0.7 (2.5) 105 |
4.2 (3.5 to 5.0) p <0.0001 |
|
Change in average daily serum [Na+] Mean (SD) N |
7.9 (4.1) 110 |
2.6 (4.2) 105 |
5.5 (4.4 to 6.5) p <0.0001 |
|
Percent of Patients Needing Fluid Restriction* |
19% 21/110 |
36% 38/106 |
p <0.01 |
|
Subgroup with Serum Sodium <125 mEq/L | |||
|
Change in average daily serum [Na+] Mean (SD) N |
5.7 (3.8) 26 |
1.0 (1.8) 30 |
5.3 (3.8 to 6.9) p <0.0001 |
|
Change in average daily serum [Na+] Mean (SD) N |
10.0 (4.8) 26 |
4.1 (4.5) 30 |
5.7 (3.1 to 8.3) p <0.0001 |
|
Percent of Patients Needing Fluid Restriction* |
35% 9/26 |
50% 15/30 |
p = 0.14 |
- Fluid Restriction defined as <1L/day at any time during treatment period.
In patients with hyponatremia (defined as <135 mEq/L), serum sodium
concentration increased to a significantly greater degree in tolvaptan-treated
patients compared to placebo-treated patients as early as 8 hours after the
first dose, and the change was maintained for 30 days. The percentage of
patients requiring fluid restriction (defined as ≤1 L/day at any time during
the treatment period) was also significantly less ( p =0.0017)
in the tolvaptan-treated group (30/215, 14%) as compared with the placebo-
treated group (51/206, 25%).
Figure 1 shows the change from baseline in serum sodium by visit in patients
with serum sodium <135 mEq/L. Within 7 days of tolvaptan discontinuation,
serum sodium concentrations in tolvaptan-treated patients declined to levels
similar to those of placebo-treated patients.
Figure 1: Pooled SALT Studies: Analysis of Mean Serum Sodium (± SD, mEq/L)
by Visit - Patients with Baseline Serum Sodium <135 mEq/L
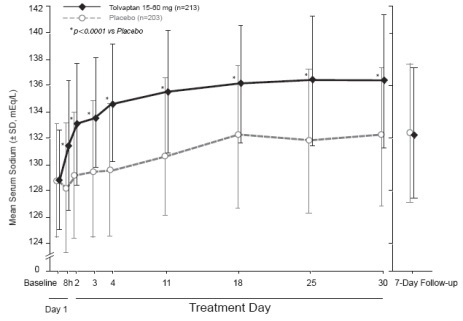
*p-value <0.0001 for all visits during tolvaptan treatment compared to placebo
Figure 2: Pooled SALT Studies: Analysis of Mean Serum Sodium (± SD, mEq/L)
by Visit - Patients with Baseline Serum Sodium <130 mEq/L
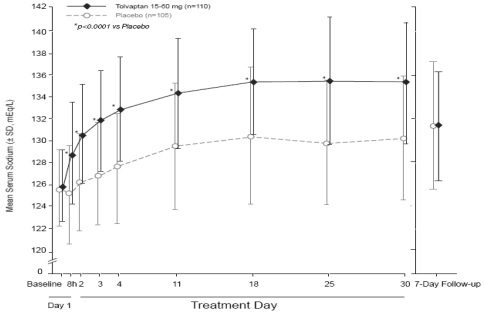
*p-value <0.0001 for all visits during tolvaptan treatment compared to placebo
In the open-label study SALTWATER, 111 patients, 94 of them hyponatremic
(serum sodium <135 mEq/L), previously on tolvaptan or placebo therapy, were
given tolvaptan as a titrated regimen (15 to 60 mg once daily) after having
returned to standard care for at least 7 days. By this time, their baseline
mean serum sodium concentration had fallen to between their original baseline
and post-placebo therapy level. Upon initiation of therapy, average serum
sodium concentrations increased to approximately the same levels as observed
for those previously treated with tolvaptan and were sustained for at least a
year. Figure 3 shows results from 111 patients enrolled in the SALTWATER
Study.
Figure 3: SALTWATER: Analysis of Mean Serum Sodium (± SD, mEq/L) by Visit
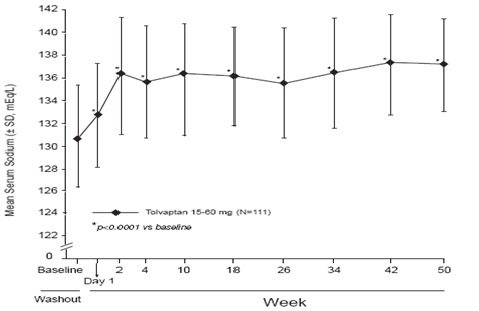
*p-value <0.0001 for all visits during tolvaptan treatment compared to baseline
14.2 Heart Failure
In a phase 3 double-blind, placebo-controlled study (EVEREST), 4133 patients with worsening heart failure were randomized to tolvaptan or placebo as an adjunct to standard of care. Long-term tolvaptan treatment (mean duration of treatment of 0.75 years) had no demonstrated effect, either favorable or unfavorable, on all-cause mortality [HR (95% CI): 0.98 (0.9, 1.1)] or the combined endpoint of CV mortality or subsequent hospitalization for worsening HF [HR (95% CI): 1.0 (0.9, 1.1)].
HOW SUPPLIED SECTION
16 HOW SUPPLIED/STORAGE AND HANDLING
How Supplied
Tolvaptan tablets are available in the following strengths and packages.
Tolvaptan tablets 15 mg are white to off white colored, triangular, bevel
edged, biconvex tablets debossed with 'H' on one side and 'T9' on the other
side.
Blister Pack of 100 (10x10) unit dose tablets (Clear PVC-Alu Peelable) NDC
31722-868-01
Blister Pack of 100 (10x10) unit dose tablets (Alu-Alu) NDC 31722-868-02
Blister Pack of 10 (1x10) unit dose tablets (Clear PVC-Alu Peelable) NDC
31722-868-03
Blister Pack of 10 (1x10) unit dose tablets (Alu-Alu) NDC 31722-868-04
Tolvaptan tablets 30 mg are blue, round, bevel edged, biconvex tablets
debossed with 'H' on one side and 'T10' on the other side.
Blister Pack of 100 (10x10) unit dose tablets (Clear PVC-Alu Peelable) NDC
31722-869-01
Blister Pack of 100 (10x10) unit dose tablets (Alu-Alu) NDC 31722-869-02
Blister Pack of 10 (1x10) unit dose tablets (Clear PVC-Alu Peelable) NDC
31722-869-03
Blister Pack of 10 (1x10) unit dose tablets (Alu-Alu) NDC 31722-869-04
Storage and Handling
Store at 20º to 25ºC (68º to 77ºF) [see USP Controlled Room Temperature].
Keep out of reach of children.
INFORMATION FOR PATIENTS SECTION
17 PATIENT COUNSELING INFORMATION
As a part of patient counseling, healthcare providers must review the
tolvaptan tablets Medication Guide with every patient [see FDA-Approved Medication Guide].
Pregnancy
Advise pregnant women of the potential risk to a fetus. Advise females of
reproductive potential to inform their prescriber of a known or suspected
pregnancy [see Use in Specific Populations ( 8.1)].
Lactation
Advise patients not to breastfeed an infant if they are taking tolvaptan
tablets [see Use in Specific Populations ( 8.2)].

Manufactured for:
Camber Pharmaceuticals, Inc.
Piscataway, NJ 08854
By: Annora Pharma Pvt. Ltd.
Sangareddy - 502313, Telangana, India.
Revised: 07/2022
SPL UNCLASSIFIED SECTION
MEDICATION GUIDE
Tolvaptan Tablets
** (tol-VAP-tan******)****
Read the Medication Guide that comes with tolvaptan tablets before you take
them and each time you get a new prescription. There may be new information.
This Medication Guide does not take the place of talking to your healthcare
provider about your medical condition or your treatment. Share this important
information with members of your household.
What is the most important information I should know about tolvaptan
tablets?
1) Tolvaptan tablets may make the salt (sodium) level in your blood rise too
fast. This can increase your risk of a serious condition called osmotic
demyelination syndrome (ODS). ODS can lead to coma or death. ODS can also
cause new symptoms such as:
• trouble speaking
• swallowing trouble or feeling like food or liquid gets stuck while
swallowing
• drowsiness
• confusion
• mood changes
• trouble controlling body movement (involuntary movement) and weakness in
muscles of the arms and legs
• seizures
You or a family member should tell your healthcare provider right away if you
have any of these symptoms even if they begin later in treatment. Also tell
your healthcare provider about any other new symptoms while taking tolvaptan
tablets.
You may be more at risk for ODS if you have:
• liver disease
• not eaten enough for a long period of time (malnourished)
• very low sodium level in your blood
• been drinking large amounts of alcohol for a long period of time (chronic
alcoholism)
To lessen your risk of ODS while taking tolvaptan tablets:
•** Treatment with tolvaptan tablets should be started and re-started only in
a hospital, where the sodium levels in your blood can be checked closely.**
• Do not take tolvaptan tablets if you cannot tell if you are thirsty.
• To prevent losing too much body water (dehydration), have water available to
drink at all times while taking tolvaptan tablets. Unless your healthcare
provider tells you otherwise, drink when you are thirsty.
• If your healthcare provider tells you to keep taking tolvaptan tablets after
you leave a hospital, it is important that you do not stop and re-start
tolvaptan tablets on your own. You may need to go back to a hospital to re-
start tolvaptan tablets. Talk to your healthcare provider right away if you
stop taking tolvaptan tablets for any reason.
• It is important to stay under the care of your healthcare provider while
taking tolvaptan tablets and follow their instructions.
2) Tolvaptan tablets may cause liver problems, including life-threatening
liver failure.
****Tolvaptan tablets should not be taken for more than 30 days. Tell your
doctor right away if you develop or have worsening of any of these signs and
symptoms of liver problems:
• Loss of appetite, nausea, vomiting
• Fever, feeling unwell, unusual tiredness
• Itching
• Yellowing of the skin or the whites of the eyes (jaundice)
• Unusual darkening of the urine
• Right upper stomach area pain or discomfort
3) If you have autosomal dominant polycystic kidney disease (ADPKD), do not
use tolvaptan tablets because you should receive the medicine (tolvaptan)
through a program that ensures laboratory monitoring of your liver.
What are tolvaptan tablets?
Tolvaptan tablets are a prescription medicine used to help increase low sodium
levels in the blood, in adults with conditions such as heart failure, and
certain hormone imbalances. Tolvaptan tablets help raise salt levels in your
blood by removing extra body water as urine.
It is not known if tolvaptan tablets are safe or works in children.
Who should not take tolvaptan tablets?
Do not take tolvaptan tablets if:
• you are allergic to tolvaptan or any of the ingredients in tolvaptan
tablets. See the end of this Medication Guide for a complete list of
ingredients in tolvaptan tablets.
• the sodium level in your blood must be increased right away.
• you cannot replace fluids by drinking or you cannot feel if you are thirsty.
• you are dizzy, faint, or your kidneys are not working normally because you
have lost too much body fluid.
• you take certain medicines. These medicines could cause you to have too much
tolvaptan in your blood:
• the antibiotic medicines, clarithromycin (Biaxin, Biaxin XL) or
telithromycin (Ketek)
• the antifungal medicines, ketoconazole (Nizoral) or itraconazole (Sporanox)
• the anti-HIV medicines, ritonavir (Kaletra, Norvir), indinavir (Crixivan),
nelfinavir (Viracept), and saquinavir (Invirase)
• the antidepressant medicine, nefazodone hydrochloride
• your body is not able to make urine. Tolvaptan tablets will not help your
condition.
What should I tell my healthcare provider before taking tolvaptan tablets?
Tell your healthcare provider about all your medical conditions, including if
you:
• have kidney problems and your body cannot make urine.
• have liver problems
• cannot feel if you are thirsty. See “What is the most important information
I should know about tolvaptan tablets?”
• had an allergic reaction in the past to tolvaptan or to any of the other
ingredients of this medicine. See the end of this Medication Guide for a list
of the ingredients in tolvaptan tablets.
• are pregnant or plan to become pregnant. It is not known if tolvaptan
tablets will harm your unborn baby.
• are breast-feeding. It is not known if tolvaptan passes into your breast
milk. You and your healthcare provider should decide if you will take
tolvaptan tablets or breast-feed. You should not do both.
• are taking desmopressin (dDAVP).
Tell your healthcare provider about all the medicines you take, including
prescription and non-prescription medicines, vitamins, and herbal supplements.
Using tolvaptan tablets with certain medicines could cause you to have too
much tolvaptan in your blood. See “Who should not take tolvaptan tablets?”
Tolvaptan tablets may affect the way other medicines work and other medicines
may affect how tolvaptan tablets work.
Know the medicines you take. Keep a list of them and show it to your
healthcare provider and pharmacist when you get a new medicine.
How should I take tolvaptan tablets?
• See “What is the most important information I should know about tolvaptan
tablets?”
• Take tolvaptan tablets exactly as prescribed by your healthcare provider.
• Take tolvaptan tablets one time each day.
• You can take tolvaptan tablets with or without food.
• Do not drink grapefruit juice during treatment with tolvaptan tablets. This
could cause you to have too much tolvaptan in your blood.
• Certain medicines or illnesses may keep you from drinking fluids or may
cause you to lose too much body fluid, such as vomiting or diarrhea. If you
have these problems, call your healthcare provider right away.
• Do not miss or skip doses of tolvaptan tablets. If you miss a dose, take it
as soon as you remember. If it is near the time of the next dose, skip the
missed dose. Just take the next dose at your regular time. Do not take 2 doses
at the same time.
•If you take too much tolvaptan, call your healthcare provider right
away. If you take an overdose of tolvaptan tablets, you may need to go to a
hospital.
• If your healthcare provider tells you to stop taking tolvaptan tablets,
follow their instructions about limiting the amount of fluid you should drink.
What are the possible side effects of tolvaptan tablets?
Tolvaptan tablets can cause serious side effects including:
** • See “What is the most important information I should know about tolvaptan
tablets?”**
** • Loss of too much body fluid (dehydration)**.
Tell your healthcare provider if you:
• have vomiting or diarrhea and cannot drink normally.
• feel dizzy or faint. These may be symptoms that you have lost too much body
fluid.
Call your healthcare provider right away if you have any of these symptoms.
The most common side effects of tolvaptan tablets are:
• thirst
• dry mouth
• weakness
• constipation
• making large amounts of urine and urinating often
• increased blood sugar levels
These are not all the possible side effects of tolvaptan tablets. Talk to your
healthcare provider about any side effect that bothers you or that does not go
away while taking tolvaptan tablets.
Call your doctor for medical advice about side effects. You may report side
effects to FDA at 1-800-FDA-1088.
** How should I store tolvaptan tablets?**
Store tolvaptan tablets between 68°F to 77°F (20°C to 25°C).
Keep tolvaptan tablets and all medicines out of the reach of children.
** General Information about tolvaptan tablets**
Medicines are sometimes prescribed for purposes other than those listed in a
Medication Guide. Do not use tolvaptan tablets for a condition for which it
was not prescribed. Do not give tolvaptan tablets to other people, even if
they have the same symptoms you have. It may harm them.
This Medication Guide summarizes the most important information about
tolvaptan tablets. If you would like more information, talk with your
healthcare provider. You can ask your healthcare provider or pharmacist for
information about tolvaptan tablets that is written for healthcare
professionals. For more information about tolvaptan tablets, call Annora
Pharma Private Limited at 1-866-495-1995.
What are the ingredients in tolvaptan tablets?
Active ingredient: tolvaptan.
Inactive ingredients: corn starch, croscarmellose sodium, lactose monohydrate,
magnesium stearate, methyl alcohol, methylene chloride, microcrystalline
cellulose and povidone. 30 mg contains FD&C Blue No # 2/Indigo caramine
Aluminum Lake as colorant.
Medication Guide available at http://camberpharma.com/medication-guides
Manufactured for:
Camber Pharmaceuticals, Inc.
Piscataway, NJ 08854
By: Annora Pharma Pvt. Ltd.
Sangareddy - 502313, Telangana, India.
Revised: 07/2021
This Medication Guide has been approved by the U.S. Food and Drug Administration.
All brands listed are the trademarks of their respective owners and are not trademarks of Annora Pharma Private Limited.