
Nexlizet
These highlights do not include all the information needed to use NEXLIZET safely and effectively. See full prescribing information for NEXLIZET. NEXLIZET (bempedoic acid and ezetimibe) tablets, for oral use Initial U.S. Approval: 2020
3fa2108c-0300-47b8-9d34-f762af7c93c6
HUMAN PRESCRIPTION DRUG LABEL
Jul 24, 2025
Esperion Therapeutics, Inc.
DUNS: 029516312
Products 1
Detailed information about drug products covered under this FDA approval, including NDC codes, dosage forms, ingredients, and administration routes.
Bempedoic Acid and Ezetimibe
Product Details
FDA regulatory identification and product classification information
FDA Identifiers
Product Classification
Product Specifications
INGREDIENTS (16)
Drug Labeling Information
PACKAGE LABEL.PRINCIPAL DISPLAY PANEL
PRINCIPAL DISPLAY PANEL - 180 mg/10 mg Tablet Bottle Label
NDC 72426-818-03
Rx only
NEXLIZET®
(bempedoic acid
and ezetimibe) tablets
Contains
30 Tablets
180 mg/10 mg

INDICATIONS & USAGE SECTION
1 INDICATIONS AND USAGE
NEXLIZET, a combination of bempedoic acid and ezetimibe, is indicated:
- As an adjunct to diet, alone or in combination with other low-density lipoprotein cholesterol (LDL-C) lowering therapies, to reduce LDL-C in adults with primary hyperlipidemia, including heterozygous familial hypercholesterolemia (HeFH).
The bempedoic acid component of NEXLIZET is indicated:
- To reduce the risk of myocardial infarction and coronary revascularization in adults who are unable to take recommended statin therapy (including those not taking a statin) with:
- established cardiovascular disease (CVD), or
- a high risk for a CVD event but without established CVD.
NEXLIZET, a combination of bempedoic acid, an adenosine triphosphate citrate lyase (ACL) inhibitor, and ezetimibe, a dietary cholesterol absorption inhibitor, is indicated:
- As an adjunct to diet, alone or in combination with other low-density lipoprotein cholesterol (LDL-C) lowering therapies, to reduce LDL-C in adults with primary hyperlipidemia, including heterozygous familial hypercholesterolemia (HeFH). (1)
The bempedoic acid component of NEXLIZET is indicated:
- To reduce the risk of myocardial infarction and coronary revascularization in adults who are unable to take recommended statin therapy (including those not taking a statin) with:
- established cardiovascular disease (CVD), or
- a high risk for a CVD event but without established CVD. (1)
CONTRAINDICATIONS SECTION
4 CONTRAINDICATIONS
NEXLIZET is contraindicated in patients with a prior hypersensitivity to ezetimibe or bempedoic acid or any of the excipients in NEXLIZET [see Adverse Reactions (6.2)]. Serious hypersensitivity reactions, such as anaphylaxis, angioedema, rash and urticaria have been reported with ezetimibe or bempedoic acid.
Known hypersensitivity to ezetimibe or bempedoic acid or any of the excipients in NEXLIZET. (4, 6.2)
WARNINGS AND PRECAUTIONS SECTION
5 WARNINGS AND PRECAUTIONS
5.1 Hyperuricemia
Bempedoic acid, a component of NEXLIZET, inhibits renal tubular OAT2 and may increase blood uric acid levels [see Clinical Pharmacology (12.3)]. In the primary hyperlipidemia trials [see Clinical Studies (14.1)], 26% of bempedoic acid-treated patients with normal baseline uric acid values (versus 9.5% placebo) experienced hyperuricemia one or more times, and 3.5% of patients experienced clinically significant hyperuricemia reported as an adverse reaction (versus 1.1% placebo). Increases in uric acid levels usually occurred within the first 4 weeks of treatment initiation, persisted throughout treatment, and returned to baseline following discontinuation of treatment. After 12 weeks of treatment, the mean placebo-adjusted increase in uric acid compared to baseline was 0.8 mg/dL for patients treated with bempedoic acid. In the cardiovascular outcomes trial [see Clinical Studies (14.2)], 16.4% of bempedoic acid-treated patients experienced clinically significant hyperuricemia reported as an adverse reaction (versus 8.2% placebo).
Elevated blood uric acid may lead to the development of gout. In the primary hyperlipidemia trials, gout was reported in 1.5% of patients treated with bempedoic acid versus 0.4% of patients treated with placebo. In the cardiovascular outcomes trial, gout was reported in 3.2% of patients treated with bempedoic acid and 2.2% treated with placebo.
Advise patients to contact their healthcare provider if symptoms of hyperuricemia occur. Assess serum uric acid when clinically indicated. Monitor patients for signs and symptoms of hyperuricemia, and initiate treatment with urate-lowering drugs as appropriate.
5.2 Tendon Rupture
Bempedoic acid, a component of NEXLIZET, is associated with an increased risk of tendon rupture or injury. In the primary hyperlipidemia trials [see Clinical Studies (14.1)], tendon rupture occurred in 0.5% of patients treated with bempedoic acid versus 0% of placebo-treated patients and involved the rotator cuff (the shoulder), biceps tendon, or Achilles tendon. Tendon rupture occurred within weeks to months of starting bempedoic acid. In the cardiovascular outcomes trial [see Clinical Studies (14.2)], tendon rupture events occurred in 1.2% of bempedoic acid-treated patients versus 0.9% of placebo-treated patients. Tendon rupture may occur more frequently in patients over 60 years of age, in those taking corticosteroid or fluoroquinolone drugs, in patients with renal failure, and in patients with previous tendon disorders.
Discontinue NEXLIZET immediately if the patient experiences rupture of a tendon. Consider discontinuing NEXLIZET if the patient experiences joint pain, swelling, or inflammation. Advise patients to rest at the first sign of tendinitis or tendon rupture and to contact their healthcare provider if tendinitis or tendon rupture symptoms occur. Consider alternative therapy in patients with a history of tendon disorders or tendon rupture.
Hyperuricemia: Elevations in serum uric acid have occurred. Assess uric acid levels periodically as clinically indicated. Monitor for signs and symptoms of hyperuricemia, and initiate treatment with urate-lowering drugs as appropriate. (5.1)
Tendon Rupture: Tendon rupture has occurred. Discontinue NEXLIZET at the first sign of tendon rupture. Avoid NEXLIZET in patients who have a history of tendon disorders or tendon rupture. (5.2)
ADVERSE REACTIONS SECTION
6 ADVERSE REACTIONS
The following clinically significant adverse reactions are described elsewhere in the labeling:
- Hyperuricemia [see Warnings and Precautions (5.1)]
- Tendon Rupture [see Warnings and Precautions (5.2)]
6.1 Clinical Trials Experience
Because clinical trials are conducted under widely varying conditions, adverse reaction rates observed in the clinical trials of a drug cannot be directly compared to rates in the clinical trials of another drug and may not reflect the rates observed in clinical practice.
Bempedoic acid
The data in Table 1 reflect exposure to bempedoic acid in two placebo- controlled primary hyperlipidemia trials that included 2,009 patients treated with bempedoic acid for 52 weeks (median treatment duration of 52 weeks) [see Clinical Studies (14.1)]. The mean age for bempedoic acid-treated patients was 65 years, 29% were female, 95% were White, 3% were Black or African American, 1% were Asian, and 1% were other races; 3% identified as Hispanic or Latino ethnicity. All patients received bempedoic acid 180 mg orally once daily plus maximally tolerated statin therapy alone or in combination with other lipid- lowering therapies. At baseline, 97% of patients had CVD and about 4% had a diagnosis of HeFH. Patients on simvastatin 40 mg/day or higher were excluded from the trials.
In the primary hyperlipidemia trials, adverse reactions led to discontinuation of treatment in 11% of bempedoic acid-treated patients and 8% of placebo- treated patients. The most common reasons for bempedoic acid treatment discontinuation were muscle spasms (0.5% versus 0.3% placebo), diarrhea (0.4% versus 0.1% placebo), and pain in extremity (0.3% versus 0.0% placebo). Adverse reactions reported in at least 2% of bempedoic acid-treated patients and more frequently than in placebo-treated patients are shown in Table 1.
Table 1. Adverse Reactions (≥ 2% and greater than placebo) in Bempedoic Acid-Treated Patients with Primary Hyperlipidemia and CVD or HeFH (Trials 2 and 3)|
Adverse Reaction |
Placebo* |
Bempedoic acid* |
|---|---|---|
| ||
|
Upper respiratory tract infection |
4.0 |
4.5 |
|
Muscle spasms |
2.3 |
3.6 |
|
Hyperuricemia† |
1.1 |
3.5 |
|
Back pain |
2.2 |
3.3 |
|
Abdominal pain or discomfort† |
2.2 |
3.1 |
|
Bronchitis |
2.5 |
3.0 |
|
Pain in extremity |
1.7 |
3.0 |
|
Anemia |
1.9 |
2.8 |
|
Elevated liver enzymes† |
0.8 |
2.1 |
In the cardiovascular outcomes trial in which 7,001 patients were exposed to bempedoic acid and 6,964 patients were exposed to placebo for a median of 3.1 years [see Clinical Studies (14.2)], adverse reactions led to discontinuation of treatment in 11% of bempedoic acid-treated patients and 10% of placebo- treated patients. Adverse reactions reported in at least 2% of bempedoic acid- treated patients and more frequently than placebo are shown in Table 2.
Table 2. Adverse Reactions (≥ 2% and 0.5% greater than placebo) in Bempedoic Acid-Treated Patients with CVD or at High Risk for CVD (Trial 4)|
Adverse Reaction |
Placebo |
Bempedoic Acid |
|---|---|---|
| ||
|
Hyperuricemia* |
8 |
16 |
|
Renal impairment† |
9 |
11 |
|
Anemia |
4 |
5 |
|
Elevated liver enzymes* |
3 |
4 |
|
Muscle spasms |
3 |
4 |
|
Gout |
2 |
3 |
|
Cholelithiasis |
1 |
2 |
Other Adverse Reactions
Tendon Rupture
In the hyperlipidemia trials, tendon rupture occurred in 0.5% of bempedoic acid-treated patients versus 0% of placebo-treated patients. In the cardiovascular outcomes trial, tendon rupture events occurred in 1.2% of bempedoic acid-treated patients versus 0.9% of placebo-treated patients.
Gout
In the hyperlipidemia trials, gout occurred in 1.5% of bempedoic acid-treated patients versus 0.4% of placebo-treated patients. In the cardiovascular outcomes trial, gout occurred in 3.2% of bempedoic acid-treated patients versus 2.2% of placebo-treated patients.
Laboratory Tests
Bempedoic acid was associated with persistent changes in multiple laboratory tests that occurred within the first 4 weeks of treatment, and returned to baseline following discontinuation of treatment.
Increase in Creatinine and Blood Urea Nitrogen
In the hyperlipidemia trials, there was a mean increase in serum creatinine of 0.05 mg/dL compared to baseline with bempedoic acid at Week 12. Approximately 3.8% of patients treated with bempedoic acid had blood urea nitrogen values that doubled (versus 1.5% placebo), and about 2.2% of patients had creatinine values that increased by 0.5 mg/dL (versus 1.1% placebo). In the cardiovascular outcomes trial, 7.1% of patients had creatinine values that increased by 0.5 mg/dL (versus 5.5% placebo) and 9.5% of patients in the bempedoic acid group had BUN values that increased ≥ 2× baseline (versus 6.2% placebo).
Decrease in Hemoglobin and Leukocytes
In the hyperlipidemia trials, approximately 5.1% of patients treated with bempedoic acid (versus 2.3% placebo) had decreases in hemoglobin levels of 2 or more g/dL and below the lower limit of normal on one or more occasion. Anemia was reported in 2.8% of patients treated with bempedoic acid and 1.9% of patients treated with placebo. Approximately 9.0% of bempedoic acid-treated patients with normal baseline leukocyte count had a decrease to less than the lower limit of normal on one or more occasion (versus 6.7% placebo). Leukocyte decrease was generally asymptomatic and did not require medical intervention. In the hyperlipidemia trials, there was a small imbalance in skin or soft tissue infections, including cellulitis (0.8% versus 0.4%), but there was no imbalance in other infections.
In the cardiovascular outcomes trial, 10.8% of patients (versus 7.4% placebo) had a decrease in hemoglobin of 2 or more g/dL and below the lower limit of normal. Anemia was reported in 4.7% of patients treated with bempedoic acid and 3.9% of patients treated with placebo. There were 9.3% of bempedoic acid- treated patients with a leukocyte count below the lower limit of normal (and normal at baseline) at any point (versus 6.8% placebo).
Increase in Platelet Count
In the hyperlipidemia trials, approximately 10.1% of bempedoic acid-treated patients (versus 4.7% placebo) had increases in platelet counts of 100× 109/L or more on one or more occasion. In the cardiovascular outcomes trial, 18.6% of patients in the bempedoic acid-treated group (versus 10.2% placebo) had an increase in platelet count of 100 × 109/L or more. Platelet count increase was asymptomatic and did not result in increased risk for thromboembolic events.
Increase in Liver Enzymes
In the hyperlipidemia trials, increases in hepatic transaminases (AST and/or ALT) were observed with bempedoic acid. In most cases, the elevations were transient and resolved or improved with continued therapy or after discontinuation of therapy. Increases to more than 3× the upper limit of normal (ULN) in AST occurred in 1.4% of patients treated with bempedoic acid versus 0.4% of placebo patients, and increases to more than 5× ULN occurred in 0.4% of bempedoic acid- treated versus 0.2% of placebo-treated patients. Increases in ALT occurred with similar incidence between bempedoic acid- and placebo-treated patients. Elevations in transaminases were generally asymptomatic and not associated with elevations ≥ 2× ULN in bilirubin or with cholestasis.
In the cardiovascular outcomes trial, the incidence of repeated and confirmed ALT and/or AST >3× ULN was 1.6% in the bempedoic acid-treated group (versus 1.0% placebo). A higher percentage of patients in the bempedoic acid-treated group had hepatic enzyme elevations versus placebo (4.5% versus 3.0%, respectively).
Increase in Creatine Kinase
In the hyperlipidemia trials, approximately 1.0% of patients (versus 0.6% placebo) had elevations of CK levels of 5 or more times the normal value on one or more occasions, and 0.4% of patients (versus 0.2% placebo) had elevations of CK levels of 10 or more times.
Ezetimibe
In 10 double-blind, placebo-controlled clinical trials [see Clinical Studies (14.1)], 2,396 patients with primary hyperlipidemia (age range 9 to 86 years, 50% were female, 90% were White, 5% were Black or African American, 2% were Asian, 3% other races; 3% identified as Hispanic or Latino ethnicity) and elevated LDL-C were treated with ezetimibe 10 mg/day for a median treatment duration of 12 weeks (range 0 to 39 weeks).
Adverse reactions reported in ≥ 2% of patients treated with ezetimibe and at an incidence greater than placebo in placebo-controlled studies of ezetimibe are shown in Table 3.
Table 3. Adverse Reactions Occurring in ≥ 2% and greater than placebo in Ezetimibe-treated Patients|
Adverse Reaction |
Placebo |
Ezetimibe 10 mg |
|---|---|---|
|
Upper respiratory tract infection |
2.5 |
4.3 |
|
Diarrhea |
3.7 |
4.1 |
|
Arthralgia |
2.2 |
3.0 |
|
Sinusitis |
2.2 |
2.8 |
|
Pain in extremity |
2.5 |
2.7 |
|
Fatigue |
1.5 |
2.4 |
|
Influenza |
1.5 |
2.0 |
NEXLIZET
In a 4-arm, 12-week, randomized, double-blind, placebo-controlled, parallel group, factorial trial, 85 patients received NEXLIZET (180 mg of bempedoic acid and 10 mg of ezetimibe) once daily [see Clinical Studies (14.1)]. The mean age for NEXLIZET-treated patients was 62 years, 51% were female, 78% were White, 19% were Black or African American, 2% were Asian, and 1% were American Indian or Alaska Native; 11% identified as Hispanic or Latino ethnicity. At baseline, 61% of patients had CVD and/or a diagnosis of HeFH. All patients received NEXLIZET plus maximally tolerated statin therapy. Patients taking simvastatin 40 mg/day or higher and patients taking non-statin lipid-lowering therapy (including fibrates, niacin, bile acid sequestrants, ezetimibe, and PCSK9 inhibitors) were excluded from the trial.
Adverse reactions led to discontinuation of treatment in 8% of patients on NEXLIZET, 5% of patients on placebo, 10% of patients on bempedoic acid, and 12% of patients on ezetimibe. The most common reason for NEXLIZET treatment discontinuation was oral discomfort (2% NEXLIZET versus 0% placebo). The most commonly reported adverse reactions (incidence ≥ 3% and greater than placebo) observed with NEXLIZET, but not observed in clinical trials of bempedoic acid or ezetimibe, were urinary tract infection (5.9% NEXLIZET versus 2.4% placebo), nasopharyngitis (4.7% NEXLIZET versus 0% placebo), and constipation (4.7% NEXLIZET versus 0% placebo).
6.2 Postmarketing Experience
The following additional adverse reactions have been reported in postmarketing experience for ezetimibe and/or bempedoic acid. Because these reactions are reported voluntarily from a population of uncertain size, it is not always possible to reliably estimate their frequency or establish a causal relationship to drug exposure.
Blood Disorders: thrombocytopenia
Gastrointestinal Disorders: abdominal pain; pancreatitis; nausea
Hepatobiliary Disorders: elevations in liver transaminases, including elevations more than 5× ULN; hepatitis; cholelithiasis; cholecystitis
Immune System Disorders: Hypersensitivity reactions including: anaphylaxis, angioedema, wheezing, rash, and urticaria
Musculoskeletal Disorders: elevated creatine phosphokinase; myopathy/rhabdomyolysis
Nervous System Disorders: dizziness; paresthesia; depression; headache
Skin and Subcutaneous Tissue Disorders: erythema multiforme
- Common adverse reactions with NEXLIZET in the primary hyperlipidemia trials (incidence ≥ 2% and more frequently than placebo) were upper respiratory tract infection, muscle spasms, hyperuricemia, back pain, abdominal pain or discomfort, bronchitis, pain in extremity, anemia, elevated liver enzymes, diarrhea, arthralgia, sinusitis, fatigue, and influenza. (6.1)
- The common adverse reaction associated with bempedoic acid in the cardiovascular outcomes trial (incidence ≥ 2% and more frequently than placebo) were hyperuricemia, renal impairment, anemia, elevated liver enzymes, muscle spasms, gout, and cholelithiasis. (6.1)
To report SUSPECTED ADVERSE REACTIONS, contact Esperion at 833-377-7633 (833 ESPRMED) or FDA at 1-800-FDA-1088 or****www.fda.gov/medwatch.
DRUG INTERACTIONS SECTION
7 DRUG INTERACTIONS
No specific pharmacokinetic drug interaction studies with NEXLIZET have been conducted. Table 4 lists drug interactions with NEXLIZET that have been identified in studies with bempedoic acid or ezetimibe.
Table 4. Clinically Important Drug Interactions with NEXLIZET|
Simvastatin | |
|
Clinical Impact: |
Concomitant use of NEXLIZET with simvastatin causes an increase in simvastatin concentration and may increase the risk of simvastatin-related myopathy [see Clinical Pharmacology (12.3)]. |
|
Intervention: |
Avoid concomitant use of NEXLIZET with simvastatin greater than 20 mg. |
|
Pravastatin | |
|
Clinical Impact: |
Concomitant use of NEXLIZET with pravastatin causes an increase in pravastatin concentration and may increase the risk of pravastatin-related myopathy [see Clinical Pharmacology (12.3)]. |
|
Intervention: |
Avoid concomitant use of NEXLIZET with pravastatin greater than 40 mg. |
|
Cyclosporine | |
|
Clinical Impact: |
Concomitant use of NEXLIZET and cyclosporine increases ezetimibe and cyclosporine concentrations. The degree of increase in ezetimibe exposure may be greater in patients with severe renal insufficiency [see Clinical Pharmacology (12.3)]. |
|
Intervention: |
Monitor cyclosporine concentrations in patients receiving NEXLIZET and cyclosporine. In patients treated with cyclosporine, weigh the potential effects of the increased exposure to ezetimibe from concomitant use against the benefits of alterations in lipid levels provided by NEXLIZET. |
|
Fibrates | |
|
Clinical Impact: |
Both fenofibrate and ezetimibe may increase cholesterol excretion into the bile, leading to cholelithiasis. Coadministration of NEXLIZET with fibrates other than fenofibrate is not recommended [see Adverse Reactions (6.1)]. |
|
Intervention: |
If cholelithiasis is suspected in a patient receiving NEXLIZET and fenofibrate, gallbladder studies are indicated and alternative lipid-lowering therapy should be considered. |
|
Cholestyramine | |
|
Clinical Impact: |
Concomitant use of NEXLIZET and cholestyramine decreases ezetimibe concentration. This may result in a reduction of efficacy [see Clinical Pharmacology (12.3)]. |
|
Intervention: |
Administer NEXLIZET either at least 2 hours before or at least 4 hours after bile acid sequestrants [see Dosage and Administration (2.2)]. |
- Simvastatin: Avoid concomitant use of NEXLIZET with simvastatin greater than 20 mg. (7)
- Pravastatin: Avoid concomitant use of NEXLIZET with pravastatin greater than 40 mg. (7)
- Cyclosporine: Monitor cyclosporine concentrations. (7)
- Fibrates: If cholelithiasis is suspected in a patient receiving NEXLIZET and fenofibrate, consider alternative lipid-lowering therapy. (6.2, 7)
RECENT MAJOR CHANGES SECTION
RECENT MAJOR CHANGES
|
Indications and Usage (1) |
03/2024 |
|
Dosage and Administration (2.1, 2.2) |
03/2024 |
|
Contraindications (4) |
03/2024 |
|
Warnings and Precautions (5.1, 5.2) |
03/2024 |
CLINICAL PHARMACOLOGY SECTION
12 CLINICAL PHARMACOLOGY
12.1 Mechanism of Action
NEXLIZET contains bempedoic acid and ezetimibe. NEXLIZET reduces elevated LDL-C through inhibition of cholesterol synthesis in the liver and absorption in the intestine.
Bempedoic acid
Bempedoic acid is an adenosine triphosphate-citrate lyase (ACL) inhibitor that lowers LDL-C by inhibition of cholesterol synthesis in the liver. ACL is an enzyme upstream of 3-hydroxy-3-methyl-glutaryl-coenzyme A (HMG-CoA) reductase in the cholesterol biosynthesis pathway. Bempedoic acid and its active metabolite, ESP15228, require coenzyme A (CoA) activation by very long-chain acyl-CoA synthetase 1 (ACSVL1) to ETC-1002-CoA and ESP15228-CoA, respectively. ACSVL1 is expressed primarily in the liver. Inhibition of ACL by ETC-1002-CoA results in decreased cholesterol synthesis in the liver and lowers LDL-C in blood via upregulation of low-density lipoprotein receptors.
Ezetimibe
Ezetimibe reduces blood cholesterol by inhibiting the absorption of cholesterol by the small intestine. The molecular target of ezetimibe has been shown to be the sterol transporter, Niemann-Pick C1-Like 1 (NPC1L1), which is involved in the intestinal uptake of cholesterol and phytosterols. Ezetimibe localizes at the brush border of the small intestine and inhibits the absorption of cholesterol, leading to a decrease in the delivery of intestinal cholesterol to the liver. This causes a reduction of hepatic cholesterol stores and an increase in LDL receptors, resulting in clearance of cholesterol from the blood.
12.2 Pharmacodynamics
Administration of bempedoic acid and ezetimibe in combination with other lipid modifying agents, decreases LDL-C, non-high density lipoprotein cholesterol (non-HDL-C), apolipoprotein B (apo B), and total cholesterol (TC) in patients with hyperlipidemia.
Cardiac Electrophysiology
At a dose of 240 mg (1.3 times the approved recommended dose), bempedoic acid does not prolong the QT interval to any clinically relevant extent.
The effect of ezetimibe or NEXLIZET on QT interval has not been evaluated.
12.3 Pharmacokinetics
Absorption
NEXLIZET
The bioavailability of NEXLIZET tablets was similar relative to that from the individual tablets, coadministered. Maximum plasma concentration (Cmax) values for bempedoic acid and its active metabolite (ESP15228) were similar between formulations, but ezetimibe glucuronide and ezetimibe Cmax values were approximately 22% and 13% lower, respectively, for NEXLIZET relative to the individual tablets, coadministered. Given a similar overall extent of ezetimibe glucuronide and ezetimibe exposure (as measured by AUC), a 22% lower Cmax is unlikely to be clinically significant.
Bempedoic acid
Following single oral administration of NEXLIZET (180 mg of bempedoic acid and 10 mg of ezetimibe), mean (± SD) Cmax and AUC of bempedoic acid were 12.6 (± 2.80) µg/mL and 202 (± 43.4) µg.hr/mL, respectively; the median time to maximum concentration (Tmax) was 3.0 hours. Following multiple-dose administration of bempedoic acid monotherapy, the steady-state maximum plasma concentration (Cmax) and AUC at 180 mg/day were 20.6 ± 6.1 µg/mL and 289.0 ± 96.4 µg∙h/mL, respectively. Bempedoic acid steady-state pharmacokinetics were generally linear over a range of >60 mg to 220 mg (approximately 33% to 122% of the recommended dosage of 180 mg daily). There were no time-dependent changes in bempedoic acid pharmacokinetics following repeat administration at the recommended dosage, and bempedoic acid steady-state was achieved after 7 days. The mean accumulation ratio was approximately 2.3-fold.
The steady-state Cmax and AUC of the active metabolite (ESP15228) of bempedoic acid were 2.8 ± 0.9 µg/mL and 51.2 ± 17.2 µg∙h/mL, respectively. ESP15228 likely made a minor contribution to the overall clinical activity of bempedoic acid based on systemic exposure, relative potency, and pharmacokinetic properties.
Ezetimibe
After a single dose of NEXLIZET to fasted adults, mean ± SD ezetimibe Cmax of 3.56 ± 1.90 ng/mL were attained with a median Tmax of 5 hr. Ezetimibe- glucuronide mean Cmax values of 107 ± 46 ng/mL were achieved with a median Tmax of 1 hr. For ezetimibe monotherapy, there was no substantial deviation from dose proportionality between 5 mg and 20 mg (0.5- to 2-fold the recommended dosage). The absolute bioavailability of ezetimibe cannot be determined, as the compound is virtually insoluble in aqueous media suitable for injection.
Effect of Food
NEXLIZET
After the administration of NEXLIZET with a high-fat, high calorie breakfast in healthy subjects, the AUC for bempedoic acid and ezetimibe were comparable to the fasted state. Compared to the fasted state, the fed state resulted in 30% and 12% reductions in Cmax and 2-hour and 2.5-hour delays in median time to attain maximum concentration (Tmax) of bempedoic acid and ezetimibe, respectively. For ezetimibe glucuronide, a 12% and 42% decrease in AUC and Cmax, respectively, were observed under fed relative to fasted conditions.
This effect of food is not considered to be clinically meaningful.
Distribution
Bempedoic acid
The bempedoic acid apparent volume of distribution (V/F) was 18 L. Plasma protein binding of bempedoic acid, its glucuronide and its active metabolite, ESP15228, were 99.3%, 98.8% and 99.2%, respectively. Bempedoic acid does not partition into blood cells.
Ezetimibe
Ezetimibe and ezetimibe-glucuronide are highly bound (>90%) to human plasma proteins.
Elimination
Bempedoic acid
The steady-state clearance (CL/F) of bempedoic acid was 11.2 mL/min after once-daily dosing; renal clearance of unchanged bempedoic acid represented less than 2% of total clearance. The mean ± SD half-life for bempedoic acid in humans was 21 ± 11 hours at steady-state.
Ezetimibe
Both ezetimibe and ezetimibe-glucuronide are eliminated from plasma with a half-life of approximately 22 hours for both.
Metabolism
Bempedoic acid
The primary route of elimination for bempedoic acid is through metabolism to the acyl glucuronide. Bempedoic acid is also reversibly converted to an active metabolite (ESP15228) based on aldo-keto reductase activity observed in vitro from human liver. Mean plasma AUC metabolite/parent drug ratio for ESP15228 following repeat-dose administration was 18% and remained constant over time. Both bempedoic acid and ESP15228 are converted to inactive glucuronide conjugates in vitro by UGT2B7. Bempedoic acid, ESP15228 and their respective conjugated forms were detected in plasma with bempedoic acid accounting for the majority (46%) of the AUC0-48h and its glucuronide being the next most prevalent (30%). ESP15228 and its glucuronide represented 10% and 11% of the plasma AUC0-48h, respectively.
Ezetimibe
Ezetimibe is primarily metabolized in the small intestine and liver via glucuronide conjugation with subsequent biliary and renal excretion. Minimal oxidative metabolism has been observed in all species evaluated.
In humans, ezetimibe is rapidly metabolized to ezetimibe-glucuronide. Ezetimibe and ezetimibe- glucuronide are the major drug-derived compounds detected in plasma, constituting approximately 10% to 20% and 80% to 90% of the total drug in plasma, respectively. Plasma concentration-time profiles exhibit multiple peaks, suggesting enterohepatic recycling.
Excretion
Bempedoic acid
Following single oral administration of 240 mg of bempedoic acid (1.3 times the approved recommended dose), approximately 70% of the total dose (bempedoic acid and its metabolites) was recovered in urine, primarily as the acyl glucuronide conjugate of bempedoic acid, and approximately 30% was recovered in feces. Less than 5% of the administered dose was excreted as unchanged bempedoic acid in feces and urine combined.
Ezetimibe
Following oral administration of 14C-ezetimibe (20 mg) to human subjects, total ezetimibe (ezetimibe + ezetimibe-glucuronide) accounted for approximately 93% of the total radioactivity in plasma. Approximately 78% and 11% of the administered radioactivity were recovered in the feces and urine, respectively, over a 10-day collection period. After 48 hours, there were no detectable levels of radioactivity in the plasma.
Ezetimibe was the major component in feces, and accounted for 69% of the administered dose, while ezetimibe-glucuronide was the major component in urine and accounted for 9% of the administered dose.
Specific Populations
Patients with Renal Impairment
Bempedoic acid
No clinically significant differences in the pharmacokinetics of bempedoic acid were observed in subjects with renal impairment (mild, moderate, and severe renal impairment or renal failure) compared to those with normal renal function.
Ezetimibe
No clinically significant differences in the pharmacokinetics of ezetimibe were observed in subjects with severe renal impairment compared to those with normal renal function.
Patients with Hepatic Impairment
NEXLIZET is not recommended in patients with moderate or severe hepatic impairment due to the unknown effects of the increased exposure to ezetimibe [see Use in Specific Populations (8.7)].
Bempedoic acid
No clinically significant differences in the pharmacokinetics of bempedoic acid and its metabolite (ESP15228) were observed in subjects with mild or moderate hepatic impairment (Child-Pugh A or B) compared to those with normal hepatic function. The effect of severe hepatic impairment (Child-Pugh C) on bempedoic acid pharmacokinetics is unknown.
Ezetimibe
After a single 10 mg dose of ezetimibe, the mean AUC for total ezetimibe increased approximately 1.7-fold in patients with mild hepatic impairment (Child-Pugh score 5 to 6), compared to healthy subjects. The mean AUC values for total ezetimibe and ezetimibe increased approximately 3- to 4-fold and 5- to 6-fold, respectively, in patients with moderate (Child-Pugh score 7 to 9) or severe hepatic impairment (Child-Pugh score 10 to 15). In a 14-day, multiple-dose study (10 mg daily) in patients with moderate hepatic impairment, the mean AUC for total ezetimibe and ezetimibe increased approximately 4-fold on day 1 and day 14 compared to healthy subjects.
Other Specific Populations
Bempedoic acid
The pharmacokinetics of bempedoic acid were not affected by age, gender, race, or weight.
Ezetimibe
Geriatrics: In a multiple-dose study with ezetimibe given 10 mg once daily for 10 days, plasma concentrations for total ezetimibe were about 2-fold higher in older (≥ 65 years) healthy subjects compared to younger subjects [see Use in Specific Populations (8.5)].
Gender: In a multiple-dose study with ezetimibe given 10 mg once daily for 10 days, plasma concentrations for total ezetimibe were slightly higher (<20%) in females than in males.
Race: The pharmacokinetics of ezetimibe is not affected by race.
Drug Interaction Studies
Bempedoic acid
Cytochrome P450 Substrates
In vitro metabolic interaction studies suggest that bempedoic acid, as well as its active metabolite and glucuronide forms are not metabolized by and do not interact with cytochrome P450 enzymes.
Transporter-mediated Drug Interactions
In vitro drug interaction studies suggest bempedoic acid, as well as its active metabolite and glucuronide form, are not substrates of commonly characterized drug transporters with the exception of bempedoic acid glucuronide, which is an OAT3 substrate. Bempedoic acid weakly inhibits OAT3 at high multiples of clinically relevant concentrations, and bempedoic acid and its glucuronide weakly inhibit OATP1B1, and OATP1B3 at clinically relevant concentrations. Bempedoic acid weakly inhibits OAT2 in vitro, which is likely the mechanism responsible for minor elevations in serum creatinine and uric acid [see Adverse Reactions (6.1)].
Probenecid
Administration of bempedoic acid 180 mg with steady-state probenecid resulted in a 1.7- and a 1.2-fold increase in bempedoic acid AUC and Cmax, respectively. AUC and Cmax for bempedoic acid active metabolite (ESP15228) were increased 1.9- and 1.5-fold, respectively. These elevations are not clinically meaningful and do not impact dosing recommendations.
Statins
The pharmacokinetic interactions between bempedoic acid (at systemic exposure relevant to the indicated CVD population) and simvastatin 20 mg, atorvastatin 10 mg, pravastatin 40 mg, and rosuvastatin 10 mg were evaluated in clinical trials.
Simvastatin: Administration of simvastatin 20 mg with 240 mg of bempedoic acid or 40 mg with 180 mg of bempedoic acid in healthy subjects at steady-state resulted in approximately 2-fold (91% for 20 mg and 96% for 40 mg) and 1.5-fold (54% for 20 mg and 52% for 40 mg) increases in simvastatin acid AUC and Cmax, respectively [see Drug Interactions (7)].
Pravastatin: Administration of pravastatin 40 mg with steady-state bempedoic acid 240 mg in healthy subjects resulted in 99% (2-fold) and 104% (2-fold) increases in pravastatin acid AUC and Cmax, respectively [see Drug Interactions (7)].
Atorvastatin and Rosuvastatin: Elevations of 1.7-fold in AUC of atorvastatin, and rosuvastatin and/or their major metabolites were observed, suggesting a weak interaction. These elevations were generally within the individual statin exposures and do not impact dosing recommendations.
Warfarin
In vitro studies indicate that bempedoic acid is not an inhibitor or inducer of CYP2C9. Because warfarin is primarily eliminated through CYP2C9, its pharmacokinetics is not expected to be altered by bempedoic acid.
Other
Bempedoic acid had no effect on the pharmacokinetics of metformin or the oral contraceptive Ortho-Novum 1/35.
Ezetimibe
Ezetimibe had no significant effect on a series of probe drugs (caffeine, dextromethorphan, tolbutamide, and IV midazolam) known to be metabolized by cytochrome P450 (1A2, 2D6, 2C8/9 and 3A4) in a "cocktail" study of twelve healthy adult males. This indicates that ezetimibe is neither an inhibitor nor an inducer of these cytochrome P450 isozymes, and it is unlikely that ezetimibe will affect the metabolism of drugs that are metabolized by these enzymes.
Cyclosporine: Administration of ezetimibe with cyclosporine (75–150 mg BID) resulted in a 2.4- and a 2.9-fold increase in total ezetimibe AUC and Cmax, respectively [see Drug Interactions (7)].
Fibrates: Administration of ezetimibe with fenofibrate (200 mg QD for 14 days) resulted in a 1.48- and a 1.64-fold increase in total ezetimibe AUC and Cmax, respectively. Administration with gemfibrozil (600 mg BID for 7 days) resulted in a 1.64- and 1.91-fold increase in total ezetimibe AUC and Cmax, respectively [see Drug Interactions (7)].
Cholestyramine: Administration of ezetimibe with cholestyramine (4 g BID for 14 days) resulted in a 55% and a 4% decrease in total ezetimibe AUC and Cmax, respectively [see Drug Interactions (7)].
No clinically meaningful pharmacokinetic interaction was observed following coadministration of ezetimibe with aluminum & magnesium hydroxide combination antacid, cimetidine, glipizide, lovastatin, pravastatin, atorvastatin, rosuvastatin, fluvastatin, simvastatin, digoxin, ethinyl estradiol/levonorgestrel, and warfarin.
CLINICAL STUDIES SECTION
14 CLINICAL STUDIES
14.1 Primary Hyperlipidemia Trials in Adults
The efficacy of NEXLIZET was investigated in a single, multi-center, randomized, double-blind, placebo-controlled, parallel group trial that enrolled 301 patients with HeFH, established CVD, or multiple risk factors for CVD on maximally tolerated statin therapy.
Trial 1 (NCT03337308) was a 4-arm, 12-week trial that assessed the efficacy of NEXLIZET in 301 patients randomized 2:2:2:1 to receive either oral NEXLIZET (180 mg of bempedoic acid and 10 mg of ezetimibe) (n = 86), bempedoic acid 180 mg (n = 88), ezetimibe 10 mg (n = 86), or placebo (n = 41) once daily as add- on to maximally tolerated statin therapy. Patients were stratified by cardiovascular risk and baseline statin intensity. Patients on simvastatin 40 mg per day or higher and patients taking non-statin lipid-lowering therapy (including fibrates, niacin, bile acid sequestrants, ezetimibe, and PCSK9 inhibitors) were excluded from the trial.
Baseline Demographics and Disease Characteristics: Overall, the mean age at baseline was 64 years (range: 30 to 87 years), 50% were 65 years of age and older, 50% were female, 81% were White, 17% were Black or African American, 1% were Asian, and 1% were other races; 12% identified as Hispanic or Latino ethnicity. Sixty-two percent (62%) of patients had clinical CVD and/or a diagnosis of HeFH. The mean baseline LDL-C was 149.7 mg/dL. At the time of randomization, 65% of patients were receiving statin therapy; and 35% were receiving high intensity statin therapy.
Efficacy Results: The primary efficacy outcome measure of the study was the percent change from baseline to Week 12 in LDL-C. The difference between NEXLIZET and placebo in mean percent change in LDL-C from baseline to Week 12 was -38% (95% CI: -47%, -30%; p <0.001). High-density lipoprotein (HDL) and triglycerides (TG) were examined as exploratory endpoints and were not included in the statistical hierarchy. The difference between NEXLIZET and placebo in mean percent change from baseline to Week 12 was -5% for HDL and median percent change from baseline to Week 12 was -11% for TG. The maximum LDL-C lowering effect was observed at Week 4. For additional results see Table 5.
Table 5. Lipid Parameters in Adult Patients with HeFH, CVD or Risk Factors for CVD on Maximally Tolerated Statin Therapy (Mean % Change from Baseline to Week 12 in Trial 1)*|
LDL-C |
non-HDL-C |
apo B |
TC | |
|---|---|---|---|---|
|
apo B = apolipoprotein B; HDL-C = high-density lipoprotein cholesterol, LDL-C = low-density lipoprotein cholesterol; LS = least squares; SE = standard error; TC = total cholesterol. | ||||
|
Background statin: atorvastatin, lovastatin, pitavastatin, pravastatin, rosuvastatin, simvastatin. | ||||
| ||||
|
NEXLIZET |
-36 |
-32 |
-25 |
-26 |
|
Bempedoic acid |
-17 |
-14 |
-12 |
-12 |
|
Ezetimibe |
-23 |
-20 |
-15 |
-16 |
|
Placebo |
2 |
2 |
6 |
1 |
|
Mean Difference of NEXLIZET versus Placebo (95% CI) |
-38 (-47, -30) |
-34 (-44, -23) |
-30 (-40, -20) |
-27 (-35, -19) |
Examination of age, sex, and race subgroups did not identify differences in response to NEXLIZET among these subgroups in any of the trials.
Bempedoic Acid
Primary Hyperlipidemia
In the two primary hyperlipidemia (52-week) trials (Trials 2 and 3) that included 3,009 adult patients with HeFH or established CVD on maximally tolerated statin therapy, the difference between bempedoic acid and placebo in mean percent change in LDL-C from baseline to Week 12 was -17% to -18%. Bempedoic acid also significantly lowered non-HDL-C (-13%), apo B (-12% to -13%), and TC (-11%) compared with placebo.
Ezetimibe
Ezetimibe Added to On-going Statin Therapy: In a multicenter, double-blind, placebo-controlled, 8-week trial, 769 patients (age range 22 to 85 years, 42% were females; 90% were White, 6% were Black or African American, 1% were Asian, and 3% were other races; 2% identified as Hispanic or Latino ethnicity) with primary hyperlipidemia, known coronary heart disease or multiple cardiovascular risk factors who were already receiving statin monotherapy, but who had not met their NCEP ATP II target LDL-C goal, were randomized to receive either ezetimibe or placebo in addition to their on-going statin therapy.
Ezetimibe, added to on-going statin therapy, significantly lowered TC (-17%), LDL-C (-25%), apo B (-19%), and non-HDL-C (-23%) relative to baseline and compared with a statin administered alone. LDL-C reductions induced by ezetimibe were generally consistent across all statins.
Ezetimibe Initiated Concurrently with a Statin: In four, multicenter, double- blind, placebo-controlled, 12-week trials, in 2,382 patients (age range 18 to 87 years, 57% were female; 88% were White, 5% were Black or African American, 2% were Asian, and 5% were other races mostly identified as Hispanic or Latino ethnicity) with hyperlipidemia, ezetimibe or placebo was orally administered alone or with various doses of atorvastatin, simvastatin, pravastatin, or lovastatin. When all patients receiving ezetimibe with a statin were compared to all those receiving the corresponding statin alone, ezetimibe significantly lowered LDL-C (ezetimibe + all atorvastatin doses [-56%] versus all atorvastatin doses alone [-44%]; ezetimibe + all simvastatin doses [-51%] versus all simvastatin doses alone [-36%]; ezetimibe + all pravastatin doses [-39%] versus all pravastatin doses alone [-25%]; ezetimibe + all lovastatin doses [-40%] versus all lovastatin doses alone [-25%]). LDL-C reductions induced by ezetimibe were generally consistent across all statins.
14.2 Cardiovascular Outcomes Trial in Adults With CVD or at High Risk for
CVD
Trial 4 (NCT02993406) was a randomized, double-blind, placebo-controlled, event-driven trial in 13,970 adult patients with established CVD (70%) or at high risk for a CVD event but without CVD (30%) who were not receiving recommended statin dosages. Patients with established CVD had documented history of coronary artery disease, symptomatic peripheral arterial disease, and/or cerebrovascular atherosclerotic disease. Patients without established CVD were considered at high risk for CVD based on meeting at least one of the following criteria:
(1) Diabetes mellitus (type 1 or type 2) in females over 65 years of age or males over 60 years of age;
(2) A Reynolds Risk score > 30% or a SCORE Risk score > 7.5% over 10 years.
Reynolds risk score and SCORE risk score evaluate a 10-year risk of having a
cardiovascular (CV) event. The Reynolds risk score is based on the following
risk factors: sex, age, smoking status, systolic blood pressure, total
cholesterol, HDL cholesterol, high sensitivity C-reactive protein (hsCRP), and
familial history of CVD events. LDL-C is an additional risk factor considered
in SCORE risk score; or
(3) A coronary artery calcium score >400 Agatston units at any time in the
past.
Patients were randomized 1:1 to receive either oral bempedoic acid 180 mg per day (n = 6,992) or placebo (n = 6,978), alone or as an add on to other background lipid-lowering therapies. Background therapy could include less than low-intensity statin dosages. Overall, 95.3% of adult patients were followed until the end of the trial or death. The median follow-up duration was 3.4 years.
Baseline Demographics and Disease Characteristics
At baseline, the mean age was 66 years (range 21 to 92 years), 59% were 65 years of age and older, 15% were 75 years of age and older, 48% were female, 91% were White, 2% were Black or African American, 4% were American Indian or Alaska Native, 2% were Asian, and 1% were other races; 17% identified as Hispanic or Latino ethnicity.
Selected additional baseline characteristics included hypertension (85%), diabetes mellitus (46%), current tobacco user (22%), eGFR < 60 mL/min per 1.73 m2 (21%), and a mean body mass index of 30 kg/m2. The mean baseline LDL-C was 139 mg/dL. At baseline, 38% of patients were taking at least one lipid modifying therapy including less than low-intensity statin dosages (23%), ezetimibe (12%), or fibrates (5%). Most patients were taking at least one other CV medication including acetylsalicylic acid (57%), selective beta blockers (52%), angiotensin converting enzyme inhibitors (40%), or angiotensin receptor blockers (32%).
Efficacy Results
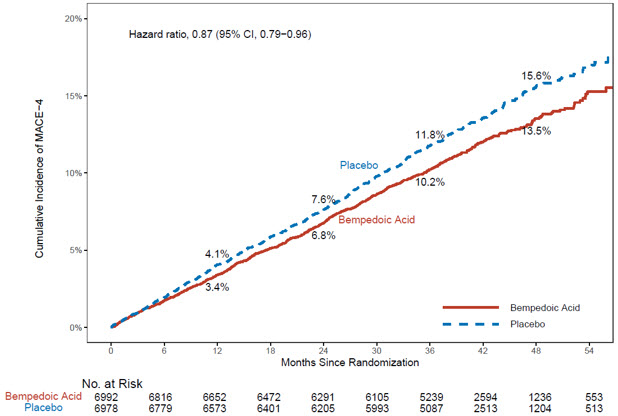
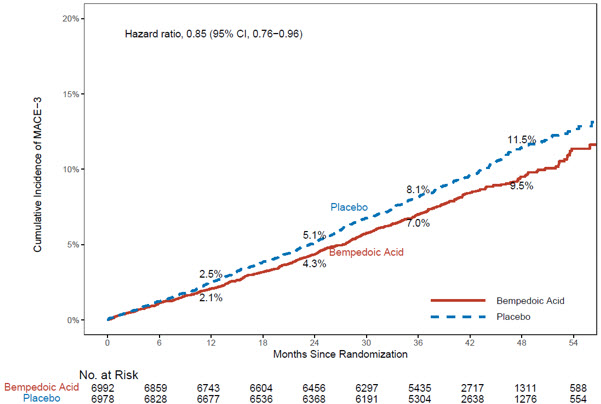
The risk for the primary composite endpoint (MACE-4: time to first occurrence of CV death, nonfatal myocardial infarction, nonfatal stroke, or coronary revascularization; p= 0.0037) and the key secondary composite endpoint (MACE-3: time to first occurrence of CV death, nonfatal myocardial infarction, or nonfatal stroke; p= 0.0058) was significantly reduced in bempedoic acid- treated patients compared to the placebo-treated patients (Table 6). The difference between bempedoic acid and placebo in mean percent change in LDL-C from baseline to Month 6 was -20% (95% CI: -21%, -19%).
Table 6: Major Cardiovascular Events in Adults with Established CVD or at High Risk for CVD (Trial 4)|
Endpoint |
Bempedoic acid |
Placebo |
Bempedoic acid vs. Placebo |
|---|---|---|---|
|
n (%) |
n (%) |
Hazard Ratio | |
|
CI = confidence interval; MACE = major adverse cardiac event. | |||
|
aHazard ratio and corresponding 95% CI were based on a Cox proportional hazard model fitting treatment as explanatory variable. | |||
|
This table also presents the time to first occurrence for each of the components of MACE; patients may have been included in more than one category. | |||
|
Primary Composite Endpoint | |||
|
Cardiovascular death, non-fatal myocardial infarction, non-fatal stroke, coronary revascularization (MACE-4) |
819 |
927 |
0.87 |
|
Key Secondary Endpoint | |||
|
Cardiovascular death, non-fatal myocardial infarction, non-fatal stroke (MACE-3) |
575 |
663 |
0.85 |
|
Components of Primary Composite Endpoint | |||
|
Non-fatal myocardial infarction |
236 |
317 |
0.73 |
|
Coronary revascularization |
435 |
529 |
0.81 |
|
Non-fatal stroke |
119 |
144 |
0.82 |
|
Cardiovascular death |
269 |
257 |
1.04 |
The Kaplan-Meier estimates of the cumulative incidence of the MACE-4 and MACE-3 endpoints are shown in Figure 1 and 2 below.
|
MACE = major adverse cardiac event |
|
Figure 1: Cumulative Incidence of Primary Composite Endpoint (MACE-4) Over 4.5 Years in Adults with Established CVD or at High Risk for CVD (Trial 4) |
|
|
|
MACE = major adverse cardiac event |
|
Figure 2: Cumulative Incidence of Composite Endpoint (MACE-3) Over 4.5 Years in Adults with Established CVD or at High Risk for CVD (Trial 4) |
|
|
INFORMATION FOR PATIENTS SECTION
17 PATIENT COUNSELING INFORMATION
Advise patients to read the FDA-approved patient labeling (Patient Information).
Risk of Hyperuricemia
Advise patients of the risk of elevated serum uric acid levels, including development of gout. Inform patients that serum uric acid levels may be monitored during treatment with NEXLIZET. Patients with signs or symptoms of hyperuricemia should contact their healthcare provider if symptoms occur [see Warnings and Precautions (5.1)].
Risk of Tendon Rupture
Inform patients of the risk of tendon rupture. Advise patients to rest at the first sign of tendinitis or tendon rupture and to immediately contact their healthcare provider if tendinitis or tendon rupture symptoms occur [see Warnings and Precautions (5.2)].
Risk of Myopathy with Concomitant Use of Simvastatin or Pravastatin
Advise patients to notify their healthcare provider(s) if they are taking, or plan to take simvastatin or pravastatin. The risk of myopathy occurring with the use of simvastatin or pravastatin may be increased when taken with NEXLIZET [see Drug Interactions (7)].
Pregnancy
Advise pregnant women of the potential risk to a fetus based on NEXLIZET's mechanism of action. Advise females to inform their healthcare provider of a known or suspected pregnancy. Advise patients that there is a pregnancy safety study that monitors pregnancy outcomes in patients exposed to NEXLIZET during pregnancy. Encourage these patients to report their pregnancy to Esperion at 1-833-377-7633 [see Use in Specific Populations (8.1)].
OVERDOSAGE SECTION
10 OVERDOSAGE
There is no clinical experience with NEXLIZET overdosage. In the event of an overdosage, consider contacting the Poison Help line (1-800-222-1222) or a medical toxicologist for additional overdosage management recommendations.
DESCRIPTION SECTION
11 DESCRIPTION
NEXLIZET tablets, for oral use, contain bempedoic acid, an adenosine triphosphate-citrate lyase (ACL) inhibitor, and ezetimibe, a dietary cholesterol absorption inhibitor.
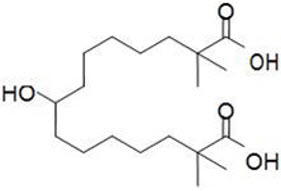
The chemical name for bempedoic acid is 8-hydroxy-2,2,14,14-tetramethyl- pentadecanedioic acid. The molecular formula is C19H36O5, and the molecular weight is 344.5 grams per mole. Bempedoic acid is a white to off-white crystalline powder that is highly soluble in ethanol, isopropanol and pH 8.0 phosphate buffer, and insoluble in water and aqueous solutions below pH 5.
Structural formula:
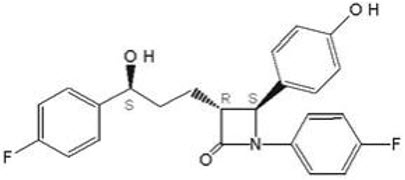
The chemical name for ezetimibe is 1-(4-fluorophenyl)-3(R)-[3-(4-fluorophenyl)-3(S)- hydroxypropyl]-4(S)-(4-hydroxyphenyl)-2-azetidinone. The molecular formula is C24H21F2NO3 and the molecular weight is 409.4 grams per mole. Ezetimibe is a white, crystalline powder that is freely to very soluble in ethanol, methanol, and acetone and practically insoluble in water.
Structural formula:
Each film-coated tablet of NEXLIZET contains 180 mg of bempedoic acid and 10 mg of ezetimibe, and the following inactive ingredients: colloidal silicon dioxide, hydroxy propyl cellulose, lactose monohydrate, magnesium stearate, microcrystalline cellulose, povidone K30, sodium lauryl sulfate, sodium starch glycolate. The film coating comprises of FD&C Blue #1/Brilliant Blue FCF Aluminum Lake, FD&C Blue #2/Indigo Carmine Aluminum Lake, glyceryl monocaprylocaprate, partially hydrolyzed polyvinyl alcohol, sodium lauryl sulfate, talc, and titanium dioxide.
NONCLINICAL TOXICOLOGY SECTION
13 NONCLINICAL TOXICOLOGY
13.1 Carcinogenesis, Mutagenesis, Impairment of Fertility
Bempedoic acid
Bempedoic acid was negative for mutagenicity in an in vitro Ames assay and negative for clastogenicity in the vitro human lymphocyte chromosome aberration assay. Bempedoic acid was negative in both in vivo mouse micronucleus and in vivo rat bone marrow micronucleus/liver comet assay. In a 2-year rat carcinogenicity study, Wistar rats were given oral doses of bempedoic acid at 3, 10 and 30 mg/kg/day. An increased incidence of liver hepatocellular adenomas and hepatocellular adenomas combined with carcinomas, thyroid gland follicular cell adenoma and follicular cell adenomas combined with carcinomas, and pancreatic islet cell adenomas combined with carcinomas were observed in male rats at the dose of 30 mg/kg/day (exposure equivalent to the maximum recommended human dose (MRHD), based on AUC). In a 2-year mice carcinogenicity study, CD-1 mice were given oral doses of bempedoic acid at 25, 75 and 150 mg/kg/day. Bempedoic acid-related increases in the incidence of liver hepatocellular adenomas, hepatocellular carcinomas and hepatocellular adenomas combined with carcinomas in male mice were observed at 75 and 150 mg/kg/day (exposures equivalent to the MRHD). Observations of liver and thyroid tumors are consistent with PPAR alpha agonism in rodents. The human relevance of pancreatic islet cell tumor findings is unknown.
In fertility and early embryofetal development study in rats, bempedoic acid was given orally to male and female rats at 10, 30 and 60 mg/kg/day. Males were dosed for 28 days prior to mating and females were dosed 14 days prior to mating through gestation day 7. No adverse effects on fertility were observed in females in the absence of maternal toxicity. No effects were observed on male fertility outcomes, but decreases in sperm counts were observed at 60 mg/kg/day (9 times the MRHD).
Ezetimibe
A 104-week dietary carcinogenicity study with ezetimibe was conducted in rats at doses up to 1,500 mg/kg/day (males) and 500 mg/kg/day (females) (approximately 20 times the human exposure at 10 mg daily based on AUC0-24hr for total ezetimibe). A 104-week dietary carcinogenicity study with ezetimibe was also conducted in mice at doses up to 500 mg/kg/day (>150 times the human exposure at 10 mg daily based on AUC0-24hr for total ezetimibe). There were no statistically significant increases in tumor incidences in drug-treated rats or mice.
No evidence of mutagenicity was observed in vitro in a microbial mutagenicity (Ames) test with Salmonella typhimurium and Escherichia coli with or without metabolic activation. No evidence of clastogenicity was observed in vitro in a chromosomal aberration assay in human peripheral blood lymphocytes with or without metabolic activation. In addition, there was no evidence of genotoxicity in the in vivo mouse micronucleus test.
In oral (gavage) fertility studies of ezetimibe conducted in rats, there was no evidence of reproductive toxicity at doses up to 1,000 mg/kg/day in male or female rats (approximately 7 times the human exposure at 10 mg daily based on AUC0-24hr for total ezetimibe).
SPL UNCLASSIFIED SECTION
Manufactured for:
Esperion Therapeutics, Inc.
3891 Ranchero Drive, Suite 150
Ann Arbor, MI 48108
NEXLIZET® (bempedoic acid and ezetimibe)
© 2025 Esperion Therapeutics, Inc.
SPL PATIENT PACKAGE INSERT SECTION
|
PATIENT INFORMATION | |||
|---|---|---|---|
|
This Patient Information has been approved by the U.S. Food and Drug Administration |
Revised: 7/2025 | ||
|
What is NEXLIZET?
It is not known if NEXLIZET is safe and effective in children. | |||
|
Do not take NEXLIZET if you are allergic to ezetimibe, bempedoic acid, or any of the ingredients in NEXLIZET. See the end of this leaflet for a complete list of ingredients in NEXLIZET. Stop taking NEXLIZET, call your healthcare provider or go to the nearest hospital emergency room right away if you have any signs or symptoms of an allergic reaction including: | |||
|
| ||
|
Before you start taking NEXLIZET, tell your healthcare provider about all your medical conditions, including if you:
NEXLIZET may affect the way other medicines work, and other medicines may
affect how NEXLIZET works.Tell your healthcare provider about all the
medicines you take, including prescription and over-the-counter medicines,
vitamins, and herbal supplements.
Know the medicines you take. Keep a list of them to show your healthcare provider and pharmacist when you get a new medicine. | |||
|
How should I take NEXLIZET?
| |||
|
What are possible side effects of NEXLIZET? *increased levels of uric acid in your blood (hyperuricemia). This can happen within 4 weeks of you starting NEXLIZET and continue throughout your treatment. Your healthcare provider may monitor your blood uric acid levels while you are taking NEXLIZET. High levels of blood uric acid may lead to gout. Call your healthcare provider if you have the following symptoms of hyperuricemia and gout: | |||
|
| ||
|
*tendon rupture or injury. Tendon problems can happen in people who take bempedoic acid, one of the medicines in NEXLIZET. Tendons are tough cords of tissue that connect muscles to bones. Symptoms of tendon problems may include pain, swelling, tears, and inflammation of tendons, most commonly with the rotator cuff (the shoulder), the biceps tendon (upper arm), and Achilles tendon at the back of the ankle. This can also happen with other tendons. Tendon ruptures can happen within weeks or months of starting NEXLIZET. *The risk of getting tendon problems while you take NEXLIZET is higher if you: | |||
|
| ||
|
*Stop taking NEXLIZET immediately and get medical help right away if you get any of the following signs or symptoms of a tendon rupture: * hear or feel a snap or pop in a tendon area * bruising right after an injury in a tendon area * unable to move the affected area or put weight on the affected area Avoid exercise and using the affected area. | |||
|
The most common side effects of NEXLIZET in people with primary hyperlipidemia include: | |||
|
| ||
|
The most common side effects of bempedoic acid in people with heart problems include: | |||
|
| ||
|
Tell your healthcare provider if you have any side effect that bothers you or
that does not go away. | |||
|
How should I store NEXLIZET?
Keep NEXLIZET and all medicines out of the reach of children. | |||
|
General information about the safe and effective use of NEXLIZET. | |||
|
What are the ingredients in NEXLIZET? *active ingredients: bempedoic acid and ezetimibe *inactive ingredients: colloidal silicon dioxide, hydroxy propyl cellulose, lactose monohydrate, magnesium stearate, microcrystalline cellulose, povidone K30, sodium lauryl sulfate, sodium starch glycolate *tablet coating: FD&C Blue #1/Brilliant Blue FCF Aluminum Lake, FD&C Blue #2/Indigo Carmine Aluminum Lake, glyceryl monocaprylocaprate, partially hydrolyzed polyvinyl alcohol, sodium lauryl sulfate, talc, and titanium dioxide | |||
|
Manufactured for: |
DOSAGE FORMS & STRENGTHS SECTION
3 DOSAGE FORMS AND STRENGTHS
NEXLIZET is available as:
- Tablets: 180 mg/10 mg, blue, oval shaped, debossed with "818" on one side and "ESP" on the other side.
Tablets: 180 mg bempedoic acid/10 mg ezetimibe (3)
DOSAGE & ADMINISTRATION SECTION
2 DOSAGE AND ADMINISTRATION
2.1 Recommended Dosage and Administration
- The recommended dosage of NEXLIZET is one tablet orally once daily. One tablet of NEXLIZET contains 180 mg of bempedoic acid and 10 mg of ezetimibe.
- Swallow the tablet whole. NEXLIZET can be taken with or without food.
- If a dose is missed, take the missed dose as soon as possible. Do not double the next dose.
- After initiation of NEXLIZET, analyze lipid levels within 8 to 12 weeks.
2.2 Coadministration with Bile Acid Sequestrants
Administer NEXLIZET either at least 2 hours before or at least 4 hours after administration of a bile acid sequestrant [see Drug Interactions (7)].
- Administer one tablet (180 mg bempedoic acid and 10 mg ezetimibe) orally once daily with or without food. (2.1)
- Swallow the tablet whole. (2.1)
- Coadministration with Bile Acid Sequestrants: Administer at least 2 hours before or at least 4 hours after bile acid sequestrants. (2.2)
USE IN SPECIFIC POPULATIONS SECTION
8 USE IN SPECIFIC POPULATIONS
8.1 Pregnancy
Risk Summary
Discontinue NEXLIZET when pregnancy is recognized unless the benefits of therapy outweigh the potential risks to the fetus.
There are insufficient data on bempedoic acid use in pregnant women to evaluate for a drug-associated risk of major birth defects, miscarriage, or adverse maternal or fetal outcomes. There are insufficient data on ezetimibe use in pregnant women to evaluate for a drug-associated risk of major birth defects, miscarriage, or adverse maternal or fetal outcomes. In animal reproduction studies, bempedoic acid was not teratogenic in rats and rabbits when administered at doses resulting in exposures up to 11 and 12 times, respectively, the human exposures at the maximum clinical dose, based on AUC. In oral (gavage) embryo-fetal development studies of ezetimibe conducted in rats and rabbits during organogenesis, there was no evidence of maternal toxicity or embryo-fetal teratogenic or toxicologic effects at exposures up to 10 and 150 times the human exposure, respectively, based on AUC (see Data). NEXLIZET decreases cholesterol synthesis and possibly the synthesis of other biologically active substances derived from cholesterol; therefore, NEXLIZET may cause fetal harm when administered to pregnant women based on the mechanism of action [see Clinical Pharmacology (12.1)]. In addition, treatment of hyperlipidemia is not generally necessary during pregnancy. Atherosclerosis is a chronic process and the discontinuation of lipid-lowering drugs during pregnancy should have little impact on the outcome of long-term therapy of primary hyperlipidemia for most patients.
The estimated background risk of major birth defects and miscarriage for the indicated population is unknown. In the U.S. general population, the estimated background risk of major birth defects and miscarriage in clinically recognized pregnancies is 2-4% and 15-20%, respectively.
Report pregnancies to the Esperion Therapeutics, Inc. Adverse Event reporting line at 1-833-377-7633.
Data
Animal Data
Bempedoic acid
Bempedoic acid was not teratogenic when given orally at doses of 60 and 80 mg/kg/day, resulting in 11 and 12 times the systemic exposure in humans at the maximum recommended human dose (MRHD) of 180 mg to pregnant rats and rabbits, respectively. In an embryofetal development study in rats, bempedoic acid was given orally to pregnant rats at 10, 30, and 60 mg/kg/day during the period of organogenesis from gestation day 6 to 17. There were increases in the incidence of non-adverse fetal skeletal variations (bent long bones and bent scapula and incomplete ossification) at doses ≥ 10 mg/kg/day (less than the clinical exposure) in the absence of maternal toxicity. At maternally toxic doses, bempedoic acid caused decreases in the numbers of viable fetuses, increases in post-implantation loss, and increased total resorptions at 60 mg/kg/day (11 times MRHD) and reduced fetal body weight at ≥ 30 mg/kg/day (4 times the MRHD). No adverse development effects were observed when bempedoic acid was given to pregnant rabbits during the period of organogenesis (gestation day 6 to 18) at doses up to 80 mg/kg/day (12 times MRHD).
In a pre- and post-natal development study in pregnant rats given oral doses of bempedoic acid at 5, 10, 20, 30 and 60 mg/kg/day throughout pregnancy and lactation (gestation day 6 to lactation day 20), there were adverse effects on delivery in the presence of maternal toxicity, including: increases in stillborn pups, reductions in numbers of live pups, pup survival, pup growth and slight delays in learning and memory at ≥ 10 mg/kg/day (at exposures equivalent to the MRHD).
Ezetimibe
In oral (gavage) embryo-fetal development studies of ezetimibe conducted in rats (gestation days 6-15) and rabbits (gestation days 7-19), there was no evidence of maternal toxicity or embryolethal effects at the doses tested (250, 500, 1,000 mg/kg/day). In rats, increased incidences of common fetal skeletal findings (extra pair of thoracic ribs, unossified cervical vertebral centra, shortened ribs) were observed at 1,000 mg/kg/day (approximately 10 times the human exposure at 10 mg daily based on AUC0-24hr for total ezetimibe). In rabbits treated with ezetimibe, an increased incidence of extra thoracic ribs was observed at 1,000 mg/kg/day (150 times the human exposure at 10 mg daily based on AUC0-24hr for total ezetimibe). The animal-to-human exposure multiple for total ezetimibe at the no-observed effect level was 6 times for rat and 134 times for rabbit.
Fetal exposure to ezetimibe (conjugated and unconjugated) was confirmed in subsequent placental transfer studies conducted using a maternal dose of 1,000 mg/kg/day. The fetal maternal plasma exposure ratio (total ezetimibe) was 1.5 for rats on gestation day 20 and 0.03 for rabbits on gestation day 22.
The effect of ezetimibe on prenatal and postnatal development and maternal function was evaluated in pregnant rats at doses of 100, 300 or 1,000 mg/kg/day from gestation day 6 through lactation day 21. No maternal toxicity or adverse developmental outcomes were observed up to and including the highest dose tested (17 times the human exposure at 10 mg daily based on AUC0-24hr for total ezetimibe).
Multiple-dose studies of ezetimibe given in combination with statins in rats and rabbits during organogenesis resulted in higher ezetimibe and statin exposures. Reproductive findings occurred at lower doses in combination therapy compared to monotherapy.
Bempedoic acid/ezetimibe fixed combination drug product (FCDP)
In a combination embryofetal development study in rats, bempedoic acid and ezetimibe were given orally at 4 and 112-times MRHD (based on AUC) during the period of organogenesis (gestation day 6 to 17) in pregnant rats. Bempedoic acid in combination with ezetimibe did not alter the effects on embryo-fetal development profile of bempedoic acid or ezetimibe.
8.2 Lactation
Risk Summary
Bempedoic acid and ezetimibe have been detected in breast milk of lactating women who received six consecutive daily doses of 180 mg bempedoic acid plus 10 mg of ezetimibe administered as a single tablet. The mean daily infant dose through breastmilk was approximately 0.03 mg/day (95% CI: 0.01; 0.06) for bempedoic acid and 0.0002 mg/day (95% CI: 0.0001; 0.0003) for ezetimibe. A mean calculated daily infant oral dosage was 0.0109 mg/kg/day for bempedoic acid and 0.0001 mg/kg/day for ezetimibe, based on a standard infant milk intake of 150 mL/kg/day. The mean (SD) relative infant dose (RID) was approximately 0.5(0.29)% for bempedoic acid and 0.04 (0.01)% for ezetimibe of the corresponding maternal weight-adjusted dosages (see Data). Concentrations of ESP15228, the active metabolite of bempedoic acid, in breast milk were below the limit of quantitation (20 ng/mL) in 7 of 8 subjects studied. Mean amounts of 0.0010 mg ezetimibe-glucuronide, the active metabolite of ezetimibe, were recovered in breast milk during the 24 hours following the final 180 mg/10 mg maternal dose. There is no information regarding the effects of NEXLIZET on the breastfed infant, or the effects on milk production.
NEXLIZET decreases cholesterol synthesis and possibly the synthesis of other biologically active substances derived from cholesterol and may cause harm to the breastfed infant. The developmental and health benefits of breastfeeding should be considered along with the mother's clinical need for NEXLIZET and any potential adverse effects on the breastfed infant from NEXLIZET or from the underlying maternal condition [see Clinical Pharmacology (12.1)].
Data
A lactation study in 8 healthy lactating women evaluated the concentrations of bempedoic acid and ezetimibe in mature breast milk. NEXLIZET 180 mg/10 mg oral tablet was given once daily for six consecutive days. The geometric mean estimates of bempedoic acid and ezetimibe Cmax in breast milk were 107.5 ng/mL (range: 56 to 234 ng/mL) and 0.630 ng/mL (range: 0.300 to 1.1 ng/mL), respectively. Maximum bempedoic acid and ezetimibe excretion occurred within 3 hours after dosing.
8.4 Pediatric Use
The safety and effectiveness of NEXLIZET have not been established in pediatric patients.
8.5 Geriatric Use
Of the 301 patients in the clinical trial of NEXLIZET, 149 (50%) were 65 years of age and older, while 49 (16%) were 75 years of age and over. No overall differences in safety or effectiveness of NEXLIZET have been observed between patients 65 years of age and older and younger adult patients.
8.7 Hepatic Impairment
No dosage adjustment is necessary in patients with mild hepatic impairment (Child-Pugh A) [see Clinical Pharmacology (12.3)]. NEXLIZET is not recommended in patients with moderate or severe hepatic impairment (Child-Pugh B or C) due to the unknown effects of the increased exposure to ezetimibe [see Clinical Pharmacology (12.3)].
- Pregnancy: Based on mechanism of action, may cause fetal harm. (8.1)
HOW SUPPLIED SECTION
16 HOW SUPPLIED/STORAGE AND HANDLING
How Supplied
NEXLIZET tablets are supplied as follows:
|
Tablet Strength |
Description |
Package Configuration |
NDC No. |
|---|---|---|---|
|
180 mg of bempedoic acid and 10 mg of ezetimibe |
blue, oval shaped, debossed with "818" on one side and "ESP" on the other side |
Bottle of 30 tablets with child-resistant cap |
72426-818-03 |
|
Bottle of 90 tablets with child-resistant cap |
72426-818-09 |
Storage and Handling
Store at 68°F to 77°F (20°C to 25°C); excursions permitted to 59°F to 86°F (15°C to 30°C) [see USP Controlled Room Temperature]. Store and dispense in the original package protected from extreme heat and humidity. Do not discard desiccant.