
Duloxetine
These highlights do not include all the information needed to use DULOXETINE DELAYED-RELEASE CAPSULES safely and effectively. See full prescribing information for DULOXETINE DELAYED-RELEASE CAPSULES. DULOXETINE delayed-release capsules, for Oral Use. Initial U.S. Approval: 2004
eb487198-a48a-47d9-bd9e-1483d9fb7ca1
HUMAN PRESCRIPTION DRUG LABEL
Feb 6, 2023
A-S Medication Solutions
DUNS: 830016429
Products 1
Detailed information about drug products covered under this FDA approval, including NDC codes, dosage forms, ingredients, and administration routes.
Duloxetine
Product Details
FDA regulatory identification and product classification information
FDA Identifiers
Product Classification
Product Specifications
INGREDIENTS (22)
Drug Labeling Information
ADVERSE REACTIONS SECTION
6 ADVERSE REACTIONS
The following serious adverse reactions are described below and elsewhere in
the labeling:
• Suicidal Thoughts and Behaviors in Children, Adolescents and Young Adults
[see Boxed Warning and Warnings and Precautions (5.1)]
• Hepatotoxicity [see Warnings and Precautions (5.2)]
• Orthostatic Hypotension, Falls and Syncope [see Warnings and Precautions (5.3)]
• Serotonin Syndrome [see Warnings and Precautions (5.4)]
• Abnormal Bleeding [see Warnings and Precautions (5.5)]
• Severe Skin Reactions [see Warnings and Precautions (5.6)]
• Discontinuation of Treatment with duloxetine delayed-release capsules [see Warnings and Precautions (5.7)]
• Activation of Mania/Hypomania [see Warnings and Precautions (5.8)]
• Angle-Closure Glaucoma [see Warnings and Precautions (5.9)]
• Seizures [see Warnings and Precautions (5.10)]
• Effect on Blood Pressure [see Warnings and Precautions (5.11)]
• Clinically Important Drug Interactions [see Warnings and Precautions (5.12)]
• Hyponatremia [see Warnings and Precautions (5.13)]
• Urinary Hesitation and Retention [see Warnings and Precautions (5.15)]
6.1 Clinical Trial Data Sources
Because clinical trials are conducted under widely varying conditions, adverse
reaction rates observed in the clinical trials of a drug cannot be directly
compared to rates in the clinical trials of another drug and may not reflect
the rates observed in practice.
The stated frequencies of adverse reactions represent the proportion of
individuals who experienced, at least once, a treatment-emergent adverse
reaction of the type listed. A reaction was considered treatment-emergent if
it occurred for the first time or worsened while receiving therapy following
baseline evaluation. Reactions reported during the studies were not
necessarily caused by the therapy, and the frequencies do not reflect
investigator impression (assessment) of causality.
Adults- The data described below reflect exposure to duloxetine in placebo-
controlled trials for MDD (N=3,779), GAD (N=1,018), OA (N=503), CLBP (N=600),
DPNP (N=906), and another indication (N=1,294). The population studied was 17
to 89 years of age; 65.7%, 60.8%, 60.6%, 42.9% and 94.4% female; and 81.8%,
72.6%, 85.3%, 74% and 85.7% Caucasian for MDD, GAD, OA and CLBP, DPNP, and
another indication, respectively. Most patients received doses of a total of
60 to 120 mg per day [see Clinical Studies (14)]. The data below do not
include results of the trial examining the efficacy of duloxetine delayed-
release capsules in patients ≥ 65 years old for the treatment of generalized
anxiety disorder; however, the adverse reactions observed in this geriatric
sample were generally similar to adverse reactions in the overall adult
population.
Children and Adolescents — The data described below reflect exposure to
duloxetine delayed-release capsules in pediatric, 10-week, placebo-controlled
trials for MDD (N=341) and GAD (N=135). The population studied (N=476) was 7
to 17 years of age with 42.4% children age 7 to 11 years of age, 50.6% female,
and 68.6% white. Patients received 30 to 120 mg per day during placebo-
controlled acute treatment studies. Additional data come from the overall
total of 822 pediatric patients (age 7 to 17 years of age) with 41.7% children
age 7 to 11 years of age and 51.8% female exposed to duloxetine delayed-
release capsules in MDD and GAD clinical trials up to 36-weeks in length, in
which most patients received 30 to 120 mg per day.
6.2 Adverse Reactions Reported as Reasons for Discontinuation of Treatment
in Adult Placebo-Controlled Trials
Major Depressive Disorder — Approximately 8.4% (319/3,779) of the patients who
received duloxetine in placebo-controlled trials for MDD discontinued
treatment due to an adverse reaction, compared with 4.6% (117/2,536) of the
patients receiving placebo. Nausea (duloxetine 1.1%, placebo 0.4%) was the
only common adverse reaction reported as a reason for discontinuation and
considered to be drug-related (i.e., discontinuation occurring in at least 1%
of the duloxetine-treated patients and at a rate of at least twice that of
placebo).
Generalized Anxiety Disorder — Approximately 13.7% (139/1,018) of the patients
who received duloxetine in placebo-controlled trials for GAD discontinued
treatment due to an adverse reaction, compared with 5% (38/767) for placebo.
Common adverse reactions reported as a reason for discontinuation and
considered to be drug-related (as defined above) included nausea (duloxetine
3.3%, placebo 0.4%), and dizziness (duloxetine 1.3%, placebo 0.4%).
Diabetic Peripheral Neuropathic Pain — Approximately 12.9% (117/906) of the
patients who received duloxetine in placebo-controlled trials for DPNP
discontinued treatment due to an adverse reaction, compared with 5.1% (23/448)
for placebo. Common adverse reactions reported as a reason for discontinuation
and considered to be drug-related (as defined above) included nausea
(duloxetine 3.5%, placebo 0.7%), dizziness (duloxetine 1.2%, placebo 0.4%),
and somnolence (duloxetine 1.1%, placebo 0%).
Chronic Pain due to Osteoarthritis — Approximately 15.7% (79/503) of the
patients who received duloxetine delayed-release capsules in 13-week, placebo-
controlled trials for chronic pain due to OA discontinued treatment due to an
adverse reaction, compared with 7.3% (37/508) for placebo. Common adverse
reactions reported as a reason for discontinuation and considered to be drug-
related (as defined above) included nausea (duloxetine 2.2%, placebo 1%).
Chronic Low Back Pain — Approximately 16.5% (99/600) of the patients who
received duloxetine delayed-release capsules in 13-week, placebo-controlled
trials for CLBP discontinued treatment due to an adverse reaction, compared
with 6.3% (28/441) for placebo. Common adverse reactions reported as a reason
for discontinuation and considered to be drug-related (as defined above)
included nausea (duloxetine delayed-release capsules 3%, placebo 0.7%), and
somnolence (duloxetine delayed-release capsules 1%, placebo 0%).
6.3 Most Common Adult Adverse Reactions
Pooled Trials for all Approved Indications — The most commonly observed
adverse reactions in duloxetine-treated patients (incidence of at least 5% and
at least twice the incidence in placebo patients) were nausea, dry mouth,
somnolence, constipation, decreased appetite, and hyperhidrosis.
Diabetic Peripheral Neuropathic Pain — The most commonly observed adverse
reactions in duloxetine-treated patients (as defined above) were nausea,
somnolence, decreased appetite, constipation, hyperhidrosis, and dry mouth.
Chronic Pain due to Osteoarthritis — The most commonly observed adverse
reactions in duloxetine-treated patients (as defined above) were nausea,
fatigue, constipation, dry mouth, insomnia, somnolence, and dizziness.
Chronic Low Back Pain — The most commonly observed adverse reactions in
duloxetine-treated patients (as defined above) were nausea, dry mouth,
insomnia, somnolence, constipation, dizziness, and fatigue.
6.4 Adverse Reactions Occurring at an Incidence of 5% or More Among
Duloxetine-Treated Patients in Adult Placebo-Controlled Trials
Table 2 gives the incidence of treatment-emergent adverse reactions in
placebo-controlled trials for approved indications that occurred in 5% or more
of patients treated with duloxetine and with an incidence greater than
placebo.
Table 2: Treatment-Emergent Adverse Reactions: Incidence of 5% or More and
Greater than Placebo in Placebo-Controlled Trials of Approved
Indications****a
|
Adverse Reaction |
Percentage of Patients Reporting Reaction | |
|
Duloxetine Delayed-Release Capsules (N=1,800) |
Placebo | |
|
Nauseac |
23 |
8 |
|
Headache |
14 |
12 |
|
Dry mouth |
13 |
5 |
|
Somnolencee |
10 |
3 |
|
Fatigueb,c |
9 |
5 |
|
Insomniad |
9 |
5 |
|
Constipationc |
9 |
4 |
|
Dizzinessc |
9 |
5 |
|
Diarrhea |
9 |
6 |
|
Decreased appetitec |
7 |
2 |
|
Hyperhidrosisc |
6 |
1 |
|
Abdominal painf |
5 |
4 |
****a The inclusion of an event in the table is determined based on the
percentages before rounding; however, the percentages displayed in the table
are rounded to the nearest integer.
b Also includes asthenia
c Events for which there was a significant dose-dependent relationship in
fixed-dose studies, excluding three MDD studies which did not have a placebo
lead-in period or dose titration.
d Also includes initial insomnia, middle insomnia, and early morning
awakening.
e Also includes hypersomnia and sedation.
f Also includes abdominal discomfort, abdominal pain lower, abdominal pain
upper, abdominal tenderness, and gastrointestinal pain.
The inclusion of an event in the table is determined based on the percentages
before rounding; however, the percentages displayed in the table are rounded
to the nearest integer. Also includes asthenia Events for which there was a
significant dose-dependent relationship in fixed-dose studies, excluding three
MDD studies which did not have a placebo lead-in period or dose titration.
Also includes initial insomnia, middle insomnia, and early morning awakening.
Also includes hypersomnia and sedation. Also includes abdominal discomfort,
abdominal pain lower, abdominal pain upper, abdominal tenderness, and
gastrointestinal pain.
The inclusion of an event in the table is determined based on the percentages before rounding; however, the percentages displayed in the table are rounded to the nearest integer. Also includes asthenia Events for which there was a significant dose-dependent relationship in fixed-dose studies, excluding three MDD studies which did not have a placebo lead-in period or dose titration. Also includes initial insomnia, middle insomnia, and early morning awakening. Also includes hypersomnia and sedation. Also includes abdominal discomfort, abdominal pain lower, abdominal pain upper, abdominal tenderness, and gastrointestinal pain.The inclusion of an event in the table is determined based on the percentages before rounding; however, the percentages displayed in the table are rounded to the nearest integer. Also includes asthenia Events for which there was a significant dose-dependent relationship in fixed-dose studies, excluding three MDD studies which did not have a placebo lead-in period or dose titration. Also includes initial insomnia, middle insomnia, and early morning awakening. Also includes hypersomnia and sedation. Also includes abdominal discomfort, abdominal pain lower, abdominal pain upper, abdominal tenderness, and gastrointestinal pain.
6.5 Adverse Reactions Occurring at an Incidence of 2% or More Among
Duloxetine-Treated Patients in Adult Placebo-Controlled Trials
Pooled MDD and GAD Trials — Table 3 gives the incidence of treatment-emergent
adverse reactions in MDD and GAD placebo-controlled trials for approved
indications that occurred in 2% or more of patients treated with duloxetine
and with an incidence greater than placebo.
Table 3: Treatment-Emergent Adverse Reactions: Incidence of 2% or More and
Greater than Placebo in MDD and GAD Placebo-Controlled Trials****a
|
System Organ Class / Adverse Reaction |
Percentage of Patients Reporting Reaction | |
|
Duloxetine Delayed-Release Capsules (N=4,797) |
Placebo | |
|
Cardiac Disorders |
2 |
1 |
|
Eye Disorders |
3 |
1 |
|
Gastrointestinal Disorders |
23 |
8 |
|
General Disorders and Administration Site Conditions |
9 |
5 |
|
Metabolism and Nutrition Disorders |
6 |
2 |
|
Nervous System Disorders |
14 |
14 |
|
Psychiatric Disorders |
9 |
5 |
|
Reproductive System and Breast Disorders |
4 |
1 |
|
Respiratory, Thoracic, and Mediastinal Disorders |
2 |
< 1 |
|
Skin and Subcutaneous TissueDisorders Hyperhidrosis |
6 |
2 |
a The inclusion of an event in the table is determined based on the
percentages before rounding; however, the percentages displayed in the table
are rounded to the nearest integer.
b For GAD, there were no adverse events that were significantly different
between treatments in adults ≥65 years that were also not significant in the
adults <65 years.
c Events for which there was a significant dose-dependent relationship in
fixed-dose studies, excluding three MDD studies which did not have a placebo
lead-in period or dose titration.
d Also includes abdominal pain upper, abdominal pain lower, abdominal
tenderness, abdominal discomfort, and gastrointestinal pain
e Also includes asthenia
f Also includes hypersomnia and sedation
g Also includes initial insomnia, middle insomnia, and early morning awakening
h Also includes feeling jittery, nervousness, restlessness, tension, and
psychomotor hyperactivity
i Also includes loss of libido
j Also includes anorgasmia
DPNP, another indication, OA, and CLBP — Table 4 gives the incidence of
treatment-emergent adverse events that occurred in 2% or more of patients
treated with duloxetine delayed-release capsules (determined prior to
rounding) in the premarketing acute phase of DPNP, another indication, OA, and
CLBP placebo-controlled trials and with an incidence greater than placebo.
Table 4: Treatment-Emergent Adverse Reactions: Incidence of 2% or More and
Greater than Placebo in DPNP, another indication, OA, and CLBP Placebo-
Controlled Trials****a
|
System Organ Class / Adverse Reaction |
Percentage of Patients Reporting Reaction | |
|
Duloxetine Delayed-Release Capsules (N=3,303) |
Placebo**(N=2,352)** | |
|
Gastrointestinal Disorders****Nausea |
23 |
7 |
|
General Disorders and Administration Site Conditions Fatigued |
11 |
5 |
|
Infections and Infestations Nasopharyngitis |
4 |
4 |
|
Metabolism and Nutrition Disorders****Decreased Appetiteb |
8 |
1 |
|
Musculo ske letal and Co nnecti ve Tissue Musculoskeletal Paine |
3 |
3 |
|
Nervous System Disorders Headache |
13 |
8 |
|
Psychiatric Disorders****Insomniab,h |
10 |
5 |
|
Reproductive System and Breast Disorders Erectile Dysfunctionb |
4 |
< 1 |
|
Respiratory, Thoracic,and Mediastinal Disorders Cough |
2 |
2 |
|
Skin and Subcutaneous Tissue Disorders |
6 |
1 |
|
Vascular Disorders****Flushingk |
3 |
1 |
a The inclusion of an event in the table is determined based on the
percentages before rounding; however, the percentages displayed in the table
are rounded to the nearest integer.
b Incidence of 120 mg/day is significantly greater than the incidence for 60
mg/day.
c Also includes abdominal discomfort, abdominal pain lower, abdominal pain
upper, abdominal tenderness and gastrointestinal pain
d Also includes asthenia
e Also includes myalgia and neck pain
f Also includes hypersomnia and sedation
g Also includes hypoaesthesia, hypoaesthesia facial, genital hypoaesthesia and
paraesthesia oral
h Also includes initial insomnia middle insomnia, and early morning awakening
i Also includes feeling jittery, nervousness, restlessness, tension and
psychomotor hyperactivity
j Also includes ejaculation failure
k Also includes hot flush
l Also includes blood pressure diastolic increased, blood pressure systolic
increased, diastolic hypertension, essential hypertension, hypertension,
hypertensive crisis, labile hypertension, orthostatic hypertension, secondary
hypertension, and systolic hypertension
6.6 Effects on Male and Female Sexual Function in Adults
Changes in sexual desire, sexual performance and sexual satisfaction often
occur as manifestations of psychiatric disorders or diabetes, but they may
also be a consequence of pharmacologic treatment. Because adverse sexual
reactions are presumed to be voluntarily underreported, the Arizona Sexual
Experience Scale (ASEX), a validated measure designed to identify sexual side
effects, was used prospectively in 4 MDD placebo-controlled trials. In these
trials, as shown in Table 5 below, patients treated with duloxetine delayed-
release capsules experienced significantly more sexual dysfunction, as
measured by the total score on the ASEX, than did patients treated with
placebo. Gender analysis showed that this difference occurred only in males.
Males treated with duloxetine delayed-release capsules experienced more
difficulty with ability to reach orgasm (ASEX Item 4) than males treated with
placebo. Females did not experience more sexual dysfunction on duloxetine
delayed-release capsules than on placebo as measured by ASEX total score.
Negative numbers signify an improvement from a baseline level of dysfunction,
which is commonly seen in depressed patients. Physicians should routinely
inquire about possible sexual side effects.
Table 5: Mean Change in ASEX Scores by Gender in MDD Placebo-Controlled
Trials
|
Male Patients****a |
Female Patients****a | |||
|
Duloxetine Delayed-Release Capsules |
Placebo (n=83) |
Duloxetine Delayed-Release Capsules (n=241) |
Placebo (n=126) | |
|
ASEX Total ( Items 1-5) Item 1 — Sex drive |
0.56b 0.40c 0.09 |
-1.07 -0.24 -0.13 |
-1.15 -0.09 -0.11 |
-1.07 -0.13 -0.17 |
a n=Number of patients with non-missing change score for ASEX total
b p=0.013 versus placebo
c p< 0.001 versus placebo
6.7 Vital Sign Changes in Adults
In placebo-controlled clinical trials across approved indications for change
from baseline to endpoint, duloxetine treatment was associated with mean
increases of 0.23 mm Hg in systolic blood pressure and 0.73 mm Hg in diastolic
blood pressure compared to mean decreases of 1.09 mm Hg systolic and 0.55 mm
Hg diastolic in placebo-treated patients. There was no significant difference
in the frequency of sustained (3 consecutive visits) elevated blood pressure
[see Warnings and Precautions (5.3, 5.11)].
Duloxetine treatment, for up to 26 weeks in placebo-controlled trials across
approved indications, typically caused a small increase in heart rate for
change from baseline to endpoint compared to placebo of up to 1.37 beats per
minute (increase of 1.20 beats per minute in dulioxetine-treated patients,
decrease of 0.17 beats per minute in placebo- treated patients).
6.8 Laboratory Changes in Adults
Duloxetine delayed-release capsules treatment in placebo-controlled clinical trials across approved indications, was associated with small mean increases from baseline to endpoint in ALT, AST, CPK, and alkaline phosphatase; infrequent, modest, transient, abnormal values were observed for these analytes in duloxetine-treated patients when compared with placebo-treated patients [see Warnings and Precautions (5.2)]. High bicarbonate, cholesterol, and abnormal (high or low) potassium, were observed more frequently in duloxetine treated patients compared to placebo.
6.9 Electrocardiogram Changes in Adults
The effect of duloxetine 160 mg and 200 mg administered twice daily to steady state was evaluated in a randomized, double-blinded, two-way crossover study in 117 healthy female subjects. No QT interval prolongation was detected. Duloxetine appears to be associated with concentration-dependent but not clinically meaningful QT shortening.
6.10 Other Adverse Reactions Observed During the Premarketing and
Postmarketing Clinical Trial Evaluation of Duloxetine in Adults
Following is a list of treatment-emergent adverse reactions reported by patients treated with duloxetine in clinical trials. In clinical trials of all indications, 34,756 patients were treated with duloxetine. Of these, 26.9% (9,337) took duloxetine for at least 6 months, and 12.4% (4,317) for at least one year. The following listing is not intended to include reactions (1) already listed in previous tables or elsewhere in labeling, (2) for which a drug cause was remote, (3) which were so general as to be uninformative, (4) which were not considered to have significant clinical implications, or (5) which occurred at a rate equal to or less than placebo.
Reactions are categorized by body system according to the following definitions: frequent adverse reactions are those occurring in at least 1/100 patients; infrequent adverse reactions are those occurring in 1/100 to 1/1,000 patients; rare reactions are those occurring in fewer than 1/1,000 patients.
Cardiac Disorders— Frequent: palpitations; Infrequent: myocardial infarction tachycardia, and Takotsubo cardiomyopathy.
Ear and Labyrinth Disorders— Frequent: vertigo; Infrequent: ear pain and tinnitus.
Endocrine Disorders— Infrequent: hypothyroidism.
Eye Disorders— Frequent: vision blurred; Infrequent: diplopia, dry eye, and visual impairment.
Gastrointestinal Disorders— Frequent: flatulence; Infrequent: dysphagia,eructation, gastritis, gastrointestinal hemorrhage, halitosis, and stomatitis; Rare: gastric ulcer.
General Disorders and Administration Site Conditions— Frequent: chills/rigors; Infrequent: falls,feeling abnormal, feeling hot and/or cold, malaise, and thirst; Rare: gait disturbance.
Infections and Infestations— Infrequent: gastroenteritis and laryngitis.
Investigations— Frequent: weight increased, weight decreased; Infrequent: blood cholesterol increased.
Metabolism and Nutrition Disorders— Infrequent: dehydration and hyperlipidemia; Rare: dyslipidemia.
Musculoskeletal and Connective Tissue Disorders— Frequent: musculoskeletal pain; Infrequent: muscle tightness and muscle twitching.
Nervous System Disorders— Frequent: dysgeusia, lethargy, and paraesthesia/hypoesthesia; Infrequent: disturbance in attention, dyskinesia, myoclonus, and poor quality sleep; Rare: dysarthria.
Psychiatric Disorders— Frequent: abnormal dreams and sleep disorder; Infrequent: apathy, bruxism, disorientation/confusional state, irritability, mood swings, and suicide attempt; Rare: completed suicide.
Renal and Urinary Disorders— Frequent: urinary frequency; Infrequent: dysuria, micturition urgency, nocturia, polyuria, and urine odor abnormal.
Reproductive System and Breast Disorders— Frequent: anorgasmia/orgasm abnormal; Infrequent: menopausal symptoms, sexual dysfunction, and testicular pain; Rare: menstrual disorder.
Respiratory, Thoracic and Mediastinal Disorders— Frequent: yawning,oropharyngeal pain; Infrequent: throat tightness.
Skin and Subcutaneous Tissue Disorders— Frequent: pruritus; Infrequent: cold sweat, dermatitis contact, erythema, increased tendency to bruise, night sweats, and photosensitivity reaction; Rare: ecchymosis.
Vascular Disorders— Frequent: hot flush; Infrequent: flushing, orthostatic hypotension, and peripheral coldness.
6.11 Adverse Reactions Observed in Children and Adolescent Placebo-
Controlled Clinical Trials
The adverse drug reaction profile observed in pediatric clinical trials
(children and adolescents) was consistent with the adverse drug reaction
profile observed in adult clinical trials. The specific adverse drug reactions
observed in adult patients can be expected to be observed in pediatric
patients (children and adolescents) [see Adverse Reactions (6.5)]. The most
common (≥ 5% and twice placebo) adverse reactions observed in pediatric
clinical trials include: nausea, diarrhea, decreased weight, and dizziness.
Table 6 provides the incidence of treatment-emergent adverse reactions in MDD
and GAD pediatric placebo-controlled trials that occurred in greater than 2%
of patients treated with duloxetine delayed-release capsules and with an
incidence greater than placebo.****
** Table 6: Treatment-Emergent Adverse Reactions: Incidence of 2% or More and
Greater than Placebo in three 10-week Pediatric Placebo-Controlled
Trials****a**
|
System Organ Class / Adverse Reaction |
** Percentageof Pediatric Patients Reporting Reaction** | |
|
Duloxetine Delayed-Release Capsules |
Placebo | |
|
Gastrointestinal Disorders |
18 |
8 |
|
General Disorders and Administration Site Conditions |
7 |
5 |
|
Investigations |
14 |
6 |
|
** Metabolism and Nutrition Disorders** |
10 |
5 |
|
** Nervous System Disorders** |
18 |
13 |
|
Psychiatric Disorders |
7 |
4 |
|
Respiratory, Thoracic, and Mediastinal Disorders |
4 |
2 |
a The inclusion of an event in the table is determined based on the
percentages before rounding; however, the percentages displayed in the table
are rounded to the nearest integer.
b Also includes abdominal pain upper, abdominal pain lower, abdominal
tenderness, abdominal discomfort, and gastrointestinal pain.
c Also includes asthenia.
d Frequency based on weight measurement meeting potentially clinically
significant threshold of ≥ 3.5% weight loss (N=467 Duloxetine delayed-release
capsules; N=354 Placebo).
e Also includes hypersomnia and sedation.
f Also includes initial insomnia, insomnia, middle insomnia, and terminal
insomnia.
Other adverse reactions that occurred at an incidence of less than 2% but were
reported by more duloxetine treated patients than placebo treated patients and
are associated duloxetine treatment: abnormal dreams (including nightmare),
anxiety, flushing (including hot flush), hyperhidrosis, palpitations, pulse
increased, and tremor.
Discontinuation-emergent symptoms have been reported when stopping duloxetine
delayed-release capsules. The most commonly reported symptoms following
discontinuation of duloxetine delayed-release capsules in pediatric clinical
trials have included headache, dizziness, insomnia, and abdominal pain [see Warnings and Precautions (5.7) and Adverse Reactions (6.2)].
Growth (Height and Weight) — Decreased appetite and weight loss have been
observed in association with the use of SSRIs and SNRIs. Pediatric patients
treated with duloxetine delayed-release capsules in clinical trials
experienced a 0.1kg mean decrease in weight at 10 weeks, compared with a mean
weight gain of approximately 0.9 kg in placebo-treated patients. The
proportion of patients who experienced a clinically significant decrease in
weight (≥3.5%) was greater in the duloxetine delayed-release capsules group
than in the placebo group (14% and 6%, respectively). Subsequently, over the
4-to 6-month uncontrolled extension periods, duloxetine delayed-release
capsules -treated patients on average trended toward recovery to their
expected baseline weight percentile based on population data from age-and sex-
matched peers. In studies up to 9 months, duloxetine-treated pediatric
patients experienced an increase in height of 1.7 cm on average (2.2 cm
increase in children [7 to 11 years of age] and 1.3 cm increase in adolescents
[12 to 17 years of age]). While height increase was observed during these
studies, a mean decrease of 1% in height percentile was observed (decrease of
2% in children [7 to 11 years of age] and increase of 0.3% in adolescents [12 to 17 years of age]). Weight and height should be monitored regularly in
children and adolescents treated with duloxetine delayed-release capsules.
6.12 Postmarketing Spontaneous Reports
The following adverse reactions have been identified during post approval use of duloxetine delayed-release capsules. Because these reactions are reported voluntarily from a population of uncertain size, it is not always possible to reliably estimate their frequency or establish a causal relationship to drug exposure.
Adverse reactions reported since market introduction that were temporally related to duloxetine therapy and not mentioned elsewhere in labeling include: acute pancreatitis, anaphylactic reaction, aggression and anger (particularly early in treatment or after treatment discontinuation), angioneurotic edema, angle-closure glaucoma, colitis (microscopic or unspecified), cutaneous vasculitis (sometimes associated with systemic involvement), extrapyramidal disorder, galactorrhea, gynecological bleeding, hallucinations, hyperglycemia, hyperprolactinemia, hypersensitivity, hypertensive crisis, muscle spasm, rash, restless legs syndrome, seizures upon treatment discontinuation, supraventricular arrhythmia, tinnitus (upon treatment discontinuation), trismus, and urticaria.
• Most common adverse reactions (≥5% and at least twice the incidence of
placebo patients): nausea, dry mouth, somnolence, constipation, decreased
appetite, and hyperhidrosis (6.3).
To report SUSPECTED ADVERSE REACTIONS, contact Hetero Labs Limited at
1-866-495-1995 or FDA at 1-800-FDA-1088 or
www.fda.gov/medwatch.
USE IN SPECIFIC POPULATIONS SECTION
8 USE IN SPECIFIC POPULATIONS
8.1 Pregnancy
Pregnancy Category C
Risk Summary — There are no adequate and well-controlled studies of duloxetine
delayed-release capsules administration in pregnant women. In animal studies
with duloxetine, fetal weights were decreased but there was no evidence of
teratogenicity in pregnant rats and rabbits at oral doses administered during
the period of organogenesis up to 4 and 7 times the maximum recommended human
dose (MRHD) of 120 mg/day, respectively. When duloxetine was administered
orally to pregnant rats throughout gestation and lactation, pup weights at
birth and pup survival to 1 day postpartum were decreased at a dose 2 times
the MRHD. At this dose, pup behaviors consistent with increased reactivity,
such as increased startle response to noise and decreased habituation of
locomotor activity were observed. Post-weaning growth was not adversely
affected. Duloxetine should be used in pregnancy only if the potential benefit
justifies the potential risk to the fetus.
Clinical Considerations
Fetal/Neonatal Adverse Reaction — Neonates exposed during pregnancy to
serotonin - norepinephrine reuptake inhibitors (SNRIs) or selective serotonin
reuptake inhibitors (SSRIs) have developed complications requiring prolonged
hospitalization, respiratory support, and tube feeding which can arise
immediately upon delivery. Reported clinical findings have included
respiratory distress, cyanosis, apnea, seizures, temperature instability,
feeding difficulty, vomiting, hypoglycemia, hypotonia, hypertonia,
hyperreflexia, tremor, jitteriness, irritability, and constant crying. These
features are consistent with either a direct toxic effect of the SNRIs or
SSRIs, or possibly, a drug discontinuation syndrome. It should be noted that,
in some cases, the clinical picture is consistent with serotonin syndrome [see Warnings and Precautions (5.4)].
Data
Animal Data — In animal reproduction studies, duloxetine has been shown to
have adverse effects on embryo/fetal and postnatal development.
When duloxetine was administered orally to pregnant rats and rabbits during
the period of organogenesis, there was no evidence of teratogenicity at doses
up to 45 mg/kg/day (4 times the maximum recommended human dose (MRHD) of 120
mg/day on a mg/m2basis, in rat; 7 times the MRHD in rabbit). However, fetal
weights were decreased at this dose, with a no-effect dose of 10mg/kg/day
approximately equal to the MRHD in rats; 2 times the MRHD in rabbits).
When duloxetine was administered orally to pregnant rats throughout gestation
and lactation, the survival of pups to 1 day postpartum and pup body weights
at birth and during the lactation period were decreased at a dose of 30
mg/kg/day (2 times the MRHD); the no-effect dose was 10 mg/kg/day.
Furthermore, behaviors consistent with increased reactivity, such as increased
startle response to noise and decreased habituation of locomotor activity,
were observed in pups following maternal exposure to 30 mg/kg/day. Post-
weaning growth and reproductive performance of the progeny were not affected
adversely by maternal duloxetine treatment.
8.3 Nursing Mothers
Risk Summary
Duloxetine is present in human milk. In a published study, lactating women who
were weaning their infants were given duloxetine. At steady state, the
concentration of duloxetine in breast milk was approximately 25% that of
maternal plasma. The estimated daily infant dose was approximately 0.14% of
the maternal dose. The developmental and health benefits of human milk feeding
should be considered along with the mother’s clinical need for duloxetine and
any potential adverse effects on the milk-fed child from the drug or from the
underlying maternal condition. Exercise caution when duloxetine is
administered to a nursing woman.
Data
The disposition of duloxetine was studied in 6 lactating women who were at
least 12 weeks postpartum and had elected to wean their infants. The women
were given 40 mg of duloxetine delayed-release capsules twice daily for 3.5
days. The peak concentration measured in breast milk occurred at a median of 3
hours after the dose. The amount of duloxetine in breast milk was
approximately 7 mcg/day while on that dose; the estimated daily infant dose
was approximately 2 mcg/kg/day. The presence of duloxetine metabolites in
breast milk was not examined.
8.4 Pediatric Use
Generalized Anxiety Disorder — In pediatric patients aged 7 to 17 years,
efficacy was demonstrated in one 10-week, placebo-controlled trial. The study
included 272 pediatric patients with GAD of which 47% were 7 to 11 years of
age. Duloxetine delayed-release capsules demonstrated superiority over placebo
as measured by greater improvement in the Pediatric Anxiety Rating Scale
(PARS) for GAD severity score [see Clinical Studies (14.2)]. The safety and
effectiveness in pediatric patients less than 7 years of age have not been
established.
Major Depressive Disorder — Efficacy was not demonstrated in two 10-week,
placebo-controlled trials with 800 pediatric patients with MDD, age 7 to 17.
Neither duloxetine delayed-release capsules nor an active control (indicated
for treatment of pediatric depression) was superior to placebo. The safety and
effectiveness in pediatric patients less than 7 years of age have not been
established.
The most frequently observed adverse reactions in the clinical trials included
nausea, headache, decreased weight, and abdominal pain. Decreased appetite and
weight loss have been observed in association with the use of SSRIs and SNRIs.
Perform regular monitoring of weight and growth in children and adolescents
treated with an SNRI such as duloxetine delayed-release capsules [see Adverse Reactions (6.11)].
Use of duloxetine delayed-release capsules in a child or adolescent must
balance the potential risks with the clinical need [see Boxed Warning and Warnings and Precautions (5.1)].
Animal Data - Duloxetine administration to young rats from post-natal day 21
(weaning) through post-natal day 90 (adult) resulted in decreased body weights
that persisted into adulthood, but recovered when drug treatment was
discontinued; slightly delayed (~1.5 days) sexual maturation in females,
without any effect on fertility; and a delay in learning a complex task in
adulthood, which was not observed after drug treatment was discontinued. These
effects were observed at the high dose of 45 mg/kg/day (2 times the MRHD, for
a child); the no-effect-level was 20 mg/kg/day (≈1 times the MRHD, for a
child).
8.5 Geriatric Use
Of the 2,418 patients in premarketing clinical studies of duloxetine delayed-
release capsules for MDD, 5.9% (143) were 65 years of age or over. Of the
1,041 patients in CLBP premarketing studies, 21.2% (221) were 65 years of age
or over. Of the 487 patients in OA premarketing studies, 40.5% (197) were 65
years of age or over. Of the 1,074 patients in the DPNP premarketing studies,
33% (357) were 65 years of age or over. Of the 1,761 patients in FM
premarketing studies, 7.9% (140) were 65 years of age or over. In the MDD,
GAD, DPNP, FM, OA and CLBP studies, no overall differences in safety or
effectiveness were generally observed between these subjects and younger
subjects, and other reported clinical experience has not identified
differences in responses between the elderly and younger patients, but greater
sensitivity of some older individuals cannot be ruled out. SSRIs and SNRIs,
including duloxetine delayed-release capsules have been associated with cases
of clinically significant hyponatremia in elderly patients, who may be at
greater risk for this adverse event [see Warnings and Precautions (5.13)].
In an analysis of data from all placebo-controlled-trials, patients treated
with duloxetine delayed-release capsule reported a higher rate of falls
compared to patients treated with placebo. The increased risk appears to be
proportional to a patient’s underlying risk for falls. Underlying risk appears
to increase steadily with age. As elderly patients tend to have a higher
prevalence of risk factors for falls such as medications, medical
comorbidities and gait disturbances, the impact of increasing age by itself on
falls during treatment with duloxetine delayed-release capsule is unclear.
Falls with serious consequences including bone fractures and hospitalizations
have been reported [see Warnings and Precautions (5.3) and Adverse Reactions (6.10)].
The pharmacokinetics of duloxetine after a single dose of 40 mg were compared
in healthy elderly females (65 to 77 years) and healthy middle-age females (32
to 50 years). There was no difference in the Cmax, but the AUC of duloxetine
was somewhat (about 25%) higher and the half-life about 4 hours longer in the
elderly females. Population pharmacokinetic analyses suggest that the typical
values for clearance decrease by approximately 1% for each year of age between
25 to 75 years of age; but age as a predictive factor only accounts for a
small percentage of between-patient variability. Dosage adjustment based on
the age of the patient is not necessary.
8.6 Gender
Duloxetine’s half-life is similar in men and women. Dosage adjustment based on gender is not necessary.
8.7 Smoking Status
Duloxetine bioavailability (AUC) appears to be reduced by about one-third in smokers. Dosage modifications are not recommended for smokers.
8.8 Race
No specific pharmacokinetic study was conducted to investigate the effects of race.
8.9 Hepatic Impairment
Patients with clinically evident hepatic impairment have decreased duloxetine metabolism and elimination. After a single 20 mg dose of duloxetine delayed- release capsules, 6 cirrhotic patients with moderate liver impairment (Child- Pugh Class B) had a mean plasma duloxetine clearance about 15% that of age- and gender-matched healthy subjects, with a 5-fold increase in mean exposure (AUC). Although Cmax was similar to normals in the cirrhotic patients, the half-life was about 3 times longer [see Dosage and Administration (2.6) and Warnings and Precautions (5.14)].
8.10 Severe Renal Impairment
Limited data are available on the effects of duloxetine in patients with end- stage renal disease (ESRD). After a single 60 mg dose of duloxetine, Cmax and AUC values were approximately 100% greater in patients with end-stage renal disease receiving chronic intermittent hemodialysis than in subjects with normal renal function. The elimination half-life, however, was similar in both groups. The AUCs of the major circulating metabolites, 4-hydroxy duloxetine glucuronide and 5-hydroxy, 6-methoxy duloxetine sulfate, largely excreted in urine, were approximately 7- to 9-fold higher and would be expected to increase further with multiple dosing. Population PK analyses suggest that mild to moderate degrees of renal impairment (estimated CrCl 30 to 80 mL/min) have no significant effect on duloxetine apparent clearance [see Dosage and Administration (2.6) and Warnings and Precautions (5.14)].
• Pregnancy: Based on animal data may cause fetal harm (8.1)
• Nursing Mothers: Exercise caution when administered to a nursing woman (8.3)
**See 17 for PATIENT COUNSELING INFORMATION and FDA-approved Medication Guide.
**
DRUG ABUSE AND DEPENDENCE SECTION
9 DRUG ABUSE AND DEPENDENCE
9.2 Abuse
In animal studies, duloxetine did not demonstrate barbiturate-like
(depressant) abuse potential.
While duloxetine delayed-release capsules have not been systematically studied
in humans for its potential for abuse, there was no indication of drug-seeking
behavior in the clinical trials. However, it is not possible to predict on the
basis of premarketing experience the extent to which a CNS active drug will be
misused, diverted, and/or abused once marketed. Consequently, physicians
should carefully evaluate patients for a history of drug abuse and follow such
patients closely, observing them for signs of misuse or abuse of duloxetine
delayed-release capsules (e.g., development of tolerance, incrementation of
dose, drug-seeking behavior).
9.3 Dependence
In drug dependence studies, duloxetine did not demonstrate dependence- producing potential in rats.
OVERDOSAGE SECTION
10 OVERDOSAGE
10.1 Signs and Symptoms
In postmarketing experience, fatal outcomes have been reported for acute overdoses, primarily with mixed overdoses, but also with duloxetine only, at doses as low as 1,000 mg. Signs and symptoms of overdose (duloxetine alone or with mixed drugs) included somnolence, coma, serotonin syndrome, seizures, syncope, tachycardia, hypotension, hypertension, and vomiting.
10.2 Management of Overdose
There is no specific antidote to duloxetine delayed-release capsules, but if
serotonin syndrome ensues, specific treatment (such as with cyproheptadine
and/or temperature control) may be considered. In case of acute overdose,
treatment should consist of those general measures employed in the management
of overdose with any drug.
An adequate airway, oxygenation, and ventilation should be assured, and
cardiac rhythm and vital signs should be monitored. Induction of emesis is not
recommended. Gastric lavage with a large-bore orogastric tube with appropriate
airway protection, if needed, may be indicated if performed soon after
ingestion or in symptomatic patients.
Activated charcoal may be useful in limiting absorption of duloxetine from the
gastrointestinal tract. Administration of activated charcoal has been shown to
decrease AUC and Cmax by an average of one-third, although some subjects had a
limited effect of activated charcoal. Due to the large volume of distribution
of this drug, forced diuresis, dialysis, hemoperfusion, and exchange
transfusion are unlikely to be beneficial.
In managing overdose, the possibility of multiple drug involvement should be
considered. A specific caution involves patients who are taking or have
recently taken duloxetine delayed-release capsules and might ingest excessive
quantities of a TCA. In such a case, decreased clearance of the parent
tricyclic and/or its active metabolite may increase the possibility of
clinically significant sequelae and extend the time needed for close medical
observation [see Warnings and Precautions (5.4) and Drug Interactions (7)].
The physician should consider contacting a poison control center
(1-800-222-1222 or www.poison.org) for additional information on the treatment
of any overdose. Telephone numbers for certified poison control centers are
listed in the Physicians' Desk Reference (PDR).
DESCRIPTION SECTION
11 DESCRIPTION
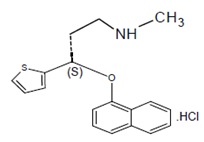
Duloxetine hydrochloride, USP is a selective serotonin and norepinephrine reuptake inhibitor (SSNRI) for oral administration. Its chemical designation is (γS)-N-Methyl-γ-(1-napthalenyloxy)-2-thiophenepropanamine hydrochloride. The empirical formula is C18H19NOS•HCl, which corresponds to a molecular weight of 333.87. The structural formula is:
Duloxetine hydrochloride, USP is an off-white to white colored crystalline powder which is freely soluble in methanol and sparingly soluble in water
Each capsule contains film-coated pellets of 22.4, 33.7, or 67.3 mg of duloxetine hydrochloride, USP equivalent to 20, 30, or 60 mg of duloxetine, respectively. Inactive ingredients include carboxy methyl ethyl cellulose, crospovidone, FD & C Blue 2, gelatin, hypromellose, isopropyl alcohol, polyethylene glycol, polysorbate 80, povidone, sodium lauryl sulfate, sucrose, sugar spheres, talc and titanium dioxide. In addition, the 20 mg and 60 mg capsules also contain iron oxide yellow.
The imprinting ink contains, butyl alcohol, dehydrated alcohol, isopropyl alcohol, propylene glycol, shellac, and strong ammonia solution. The 20 mg capsule also contains black iron oxide and potassium hydroxide. The 30 mg capsule also contains yellow iron oxide. The 60 mg capsule also contains potassium hydroxide and titanium dioxide.
CLINICAL PHARMACOLOGY SECTION
12 CLINICAL PHARMACOLOGY
12.1 Mechanism of Action
Although the exact mechanisms of the antidepressant, central pain inhibitory and anxiolytic actions of duloxetine in humans are unknown, these actions are believed to be related to its potentiation of serotonergic and noradrenergic activity in the CNS.
12.2 Pharmacodynamics
Preclinical studies have shown that duloxetine is a potent inhibitor of
neuronal serotonin and norepinephrine reuptake and a less potent inhibitor of
dopamine reuptake. Duloxetine has no significant affinity for dopaminergic,
adrenergic, cholinergic, histaminergic, opioid, glutamate, and GABA receptors
in vitro. Duloxetine does not inhibit monoamine oxidase (MAO).
Duloxetine delayed-release capsules are in a class of drugs known to affect
urethral resistance. If symptoms of urinary hesitation develop during
treatment with duloxetine delayed-release capsules, consideration should be
given to the possibility that they might be drug-related.
12.3 Pharmacokinetics
Duloxetine has an elimination half-life of about 12 hours (range 8 to 17
hours) and its pharmacokinetics are dose proportional over the therapeutic
range. Steady-state plasma concentrations are typically achieved after 3 days
of dosing. Elimination of duloxetine is mainly through hepatic metabolism
involving two P450 isozymes, CYP1A2 and CYP2D6.
Absorption and Distribution — Orally administered duloxetine hydrochloride is
well absorbed. There is a median 2 hour lag until absorption begins (Tlag),
with maximal plasma concentrations (Cmax) of duloxetine occurring 6 hours post
dose. Food does not affect the Cmax of duloxetine, but delays the time to
reach peak concentration from 6 to 10 hours and it marginally decreases the
extent of absorption (AUC) by about 10%. There is a 3 hour delay in absorption
and a one-third increase in apparent clearance of duloxetine after an evening
dose as compared to a morning dose.
The apparent volume of distribution averages about 1,640 L. Duloxetine is
highly bound (> 90%) to proteins in human plasma, binding primarily to albumin
and α1-acid glycoprotein. The interaction between duloxetine and other highly
protein bound drugs has not been fully evaluated. Plasma protein binding of
duloxetine is not affected by renal or hepatic impairment.
Metabolism and Elimination — Biotransformation and disposition of duloxetine
in humans have been determined following oral administration of 14C-labeled
duloxetine. Duloxetine comprises about 3% of the total radiolabeled material
in the plasma, indicating that it undergoes extensive metabolism to numerous
metabolites. The major biotransformation pathways for duloxetine involve
oxidation of the naphthyl ring followed by conjugation and further oxidation.
Both CYP1A2 and CYP2D6 catalyze the oxidation of the naphthyl ring in vitro.
Metabolites found in plasma include 4-hydroxy duloxetine glucuronide and
5-hydroxy, 6-methoxy duloxetine sulfate. Many additional metabolites have been
identified in urine, some representing only minor pathways of elimination.
Only trace (< 1% of the dose) amounts of unchanged duloxetine are present in
the urine. Most (about 70%) of the duloxetine dose appears in the urine as
metabolites of duloxetine; about 20% is excreted in the feces. Duloxetine
undergoes extensive metabolism, but the major circulating metabolites have not
been shown to contribute significantly to the pharmacologic activity of
duloxetine.
Children and Adolescents (ages 7 to 17 years) — Duloxetine steady-state plasma
concentration was comparable in children (7 to 12 years of age), adolescents
(13 to 17 years of age) and adults. The average steady-state duloxetine
concentration was approximately 30% lower in the pediatric population
(children and adolescents) relative to the adults. The model-predicted
duloxetine steady state plasma concentrations in children and adolescents were
mostly within the concentration range observed in adult patients and did not
exceed the concentration range in adults.