
Contrave
These highlights do not include all the information needed to use CONTRAVE safely and effectively. See full prescribing information for CONTRAVE. CONTRAVE (naltrexone hydrochloride and bupropion hydrochloride) extended-release tablets, for oral use Initial U.S. Approval: 2014
485ff360-32c8-11df-928b-0002a5d5c51b
HUMAN PRESCRIPTION DRUG LABEL
Nov 30, 2023
Nalpropion Pharmaceuticals LLC
DUNS: 081341086
Products 1
Detailed information about drug products covered under this FDA approval, including NDC codes, dosage forms, ingredients, and administration routes.
naltrexone hydrochloride and bupropion hydrochloride
Product Details
FDA regulatory identification and product classification information
FDA Identifiers
Product Classification
Product Specifications
INGREDIENTS (13)
Drug Labeling Information
DOSAGE & ADMINISTRATION SECTION
2 DOSAGE AND ADMINISTRATION
2.1 Recommended Dosing
CONTRAVE dosing should be escalated according to the following schedule:
|
Morning Dose |
Evening Dose | |
|---|---|---|
|
Week 1 |
1 tablet |
None |
|
Week 2 |
1 tablet |
1 tablet |
|
Week 3 |
2 tablets |
1 tablet |
|
Week 4 – Onward |
2 tablets |
2 tablets |
A total daily dosage of two CONTRAVE 8 mg/90 mg tablets twice daily (32 mg/360 mg) is reached at the start of Week 4.
CONTRAVE should be taken by mouth in the morning and in the evening. The tablets should not be cut, chewed, or crushed. Total daily doses greater than 32 mg/360 mg per day (two tablets twice daily) are not recommended. In clinical trials, CONTRAVE was administered with meals. However, CONTRAVE should not be taken with a high-fat meal because of a resulting significant increase in bupropion and naltrexone systemic exposure [see Warnings and Precautions (5.3) and Clinical Pharmacology (12.3)].
Patients may develop elevated blood pressure or heart rate during CONTRAVE treatment; the risk may be greater during the initial three months of therapy [see Warnings and Precautions (5.6)]. Because patients with hypertension may be at increased risk for developing blood pressure elevations, such patients should be monitored for this potential effect when initiating treatment with CONTRAVE.
Response to therapy should be evaluated after 12 weeks at the maintenance dosage. If a patient has not lost at least 5% of baseline body weight, discontinue CONTRAVE, as it is unlikely that the patient will achieve and sustain clinically meaningful weight loss with continued treatment.
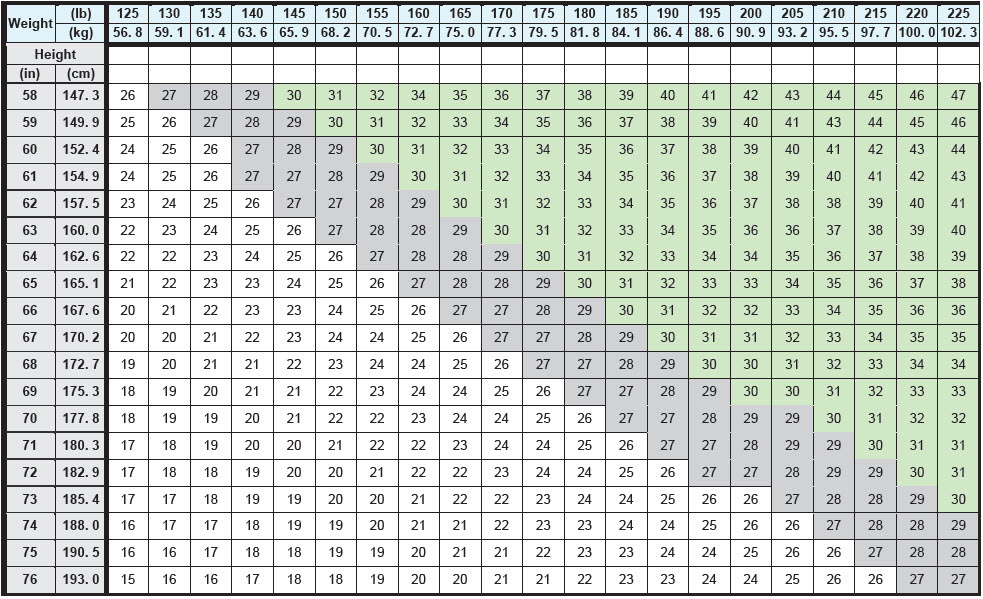
BMI is calculated by dividing weight (in kg) by height (in meters) squared. A BMI chart for determining BMI based on height and weight is provided in Table 1.
Table 1. BMI Conversion Chart
2.2 Dose Adjustment in Patients with Renal Impairment
In patients with moderate or severe renal impairment, the maximum recommended daily dose for CONTRAVE is two tablets (one tablet each morning and evening). CONTRAVE is not recommended for use in patients with end-stage renal disease [see Use in Specific Population (8.6) and Clinical Pharmacology (12.3)].
2.3 Dose Adjustment in Patients with Hepatic Impairment
In patients with moderate hepatic impairment, the maximum recommended daily maintenance dose of CONTRAVE is two tablets (one tablet each morning and evening). CONTRAVE is not recommended for use in patients with severe hepatic impairment [see Use in Specific Population (8.7) and Clinical Pharmacology (12.3)].
2.4 Switching a Patient To or From a Monoamine Oxidase Inhibitor (MAOI)
Antidepressant
At least 14 days should elapse between discontinuation of an MAOI intended to treat depression and initiation of therapy with CONTRAVE. Conversely, at least 14 days should be allowed after stopping CONTRAVE before starting an MAOI antidepressant [see Contraindications (4) and Drug Interactions (7.1)].
2.5 Concomitant Use with CYP2B6 Inhibitors
During concomitant use with CYP2B6 inhibitors (e.g., ticlopidine or clopidogrel), the maximum recommended daily dose of CONTRAVE is two tablets (one tablet each morning and evening) [see Drug Interactions (7.4) and Clinical Pharmacology (12.3)].
CONTRAVE dose escalation schedule (2.1):
|
Morning Dose |
Evening Dose | |
|---|---|---|
|
Week 1 |
1 tablet |
None |
|
Week 2 |
1 tablet |
1 tablet |
|
Week 3 |
2 tablets |
1 tablet |
|
Week 4 – Onward |
2 tablets |
2 tablets |
USE IN SPECIFIC POPULATIONS SECTION
8 USE IN SPECIFIC POPULATIONS
8.1 Pregnancy
Risk Summary
Weight loss offers no benefit to a pregnant patient and may cause fetal harm. When a pregnancy is recognized, advise the pregnant patient of the risk to the fetus, and discontinue CONTRAVE (see Clinical Considerations). Available pharmacovigilance data and data from clinical trials with the individual components of CONTRAVE use in pregnant patients have not demonstrated a drug- associated risk of major birth defects, miscarriage or adverse maternal or fetal outcomes.
Bupropion
Data from epidemiological studies of pregnant patients exposed to bupropion in the first trimester have not identified an increased risk of congenital malformations overall (see Data). When bupropion was administered to pregnant rats during organogenesis, there was no evidence of fetal malformations at doses up to approximately 20 times the maximum recommended human dose (MRHD) of 360 mg/day. When given to pregnant rabbits during organogenesis, non- dose–related increases in incidence of fetal malformations, and skeletal variations were observed at doses approximately twice the MRHD and greater. Decreased fetal weights were seen at doses 5 times the MRHD and greater (see Data).
Naltrexone
Limited case report data of pregnant patients exposed to naltrexone in the first trimester have not identified an increased risk of congenital malformations overall. Daily oral administration of naltrexone during the period of organogenesis has been shown to increase the incidence of early fetal loss in rats and rabbits at doses ≥ 15 times and ≥ 60 times the MRHD of 32 mg/day, respectively. There was no evidence of fetal malformations in rats and rabbits at doses up to approximately 100 and 200 times the MHRD, respectively. (see Data).
The estimated background risk of major birth defects and miscarriage for the indicated population is unknown. In the U.S. general population, the estimated background risk of major birth defects and miscarriage in clinically recognized pregnancies is 2-4% and 15-20%, respectively.
Clinical Considerations
Disease-associated maternal and/or embryo/fetal risk
Appropriate weight gain based on pre-pregnancy weight is currently recommended for all pregnant patients, including those who are already overweight or obese, due to the obligatory weight gain that occurs in maternal tissues during pregnancy.
Data
Human Data
In clinical studies, 21 (0.7%) of 3,024 women became pregnant while taking CONTRAVE: 11 carried to term and gave birth to a healthy infant, three had elective abortions, four had spontaneous abortions, and the outcome of three pregnancies were unknown.
Data from the international bupropion Pregnancy Registry (675 first trimester exposures) and a retrospective cohort study using the United Healthcare database (1,213 first trimester exposures) did not show an increased risk for malformations overall.
No increased risk for cardiovascular malformations overall has been observed after bupropion exposure during the first trimester. The prospectively observed rate of cardiovascular malformations in pregnancies with exposure to bupropion in the first trimester from the international Pregnancy Registry was 1.3% (9 cardiovascular malformations out of 675 first-trimester maternal bupropion exposures), which is similar to the background rate of cardiovascular malformations (approximately 1%). Data from the United Healthcare database and a case-control study (6,853 infants with cardiovascular malformations and 5,763 with non-cardiovascular malformations) from the National Birth Defects Prevention Study (NBDPS) did not show an increased risk for cardiovascular malformations overall after bupropion exposure during the first trimester.
Study findings on bupropion exposure during the first trimester and risk for left ventricular outflow tract obstruction (LVOTO) are inconsistent and do not allow conclusions regarding a possible association. The United Healthcare database lacked sufficient power to evaluate this association; the NBDPS found increased risk for LVOTO (n = 10; adjusted odds ratio [OR] = 2.6; 95% CI: 1.2, 5.7), and the Slone Epidemiology case control study did not find increased risk for LVOTO.
Study findings on bupropion exposure during the first trimester and risk for ventricular septal defect (VSD) are inconsistent and do not allow conclusions regarding a possible association. The Slone Epidemiology Study found an increased risk for VSD following first trimester maternal bupropion exposure (n = 17; adjusted OR = 2.5; 95% CI: 1.3, 5.0) but did not find increased risk for any other cardiovascular malformations studied (including LVOTO as above). The NBDPS and United Healthcare database study did not find an association between first trimester maternal bupropion exposure and VSD.
For the findings of LVOTO and VSD, the studies were limited by the small number of exposed cases, inconsistent findings among studies, and the potential for chance findings from multiple comparisons in case control studies.
Animal Data
Reproduction and developmental studies have not been conducted for the combined products naltrexone and bupropion in CONTRAVE. Separate studies with bupropion and naltrexone have been conducted in pregnant rats and rabbits. Safety margins were estimated using body surface area exposure (mg/m2) based on a body weight of 100 kg.
Daily oral administration of naltrexone has been shown to increase the incidence of early fetal loss when given to rats at doses ≥30 mg/kg/day (15 times the MHRD on a mg/m2 basis) and to rabbits at oral doses ≥60 mg/kg/day (60 times the MHRD on a mg/m2 basis).
Daily oral administration of naltrexone to rats and rabbits during the period of organogenesis did not induce malformations at doses up to 200 mg/kg/day (approximately 100 and 200 times the MHRD, respectively, on a mg/m2 basis).
Rats do not form appreciable quantities of the major human metabolite, 6-beta- naltrexol; therefore, the potential reproductive toxicity of the metabolite in rats is not known. In studies conducted in pregnant rats and rabbits, bupropion was administered orally during the period of organogenesis at doses of up to 450 and 150 mg/kg/day, respectively (approximately 20 and 14 times the MRHD, respectively, on a mg/m2 basis). There was no evidence of fetal malformations in rats. When given to pregnant rabbits during organogenesis, non-dose-related increases in incidence of fetal malformations and skeletal variations were observed at the lowest dose tested (25 mg/kg/day, approximately 2 times the MRHD on a mg/m2 basis) and greater. Decreased fetal weights were observed at doses of 50 mg/kg/day (approximately 5 times the MRHD on a mg/m2 basis) and greater. No maternal toxicity was evident at doses of 50 mg/kg/day or less.
In a pre- and postnatal development study, bupropion administered orally to pregnant rats at doses of up to 150 mg/kg/day (approximately 7 times the MRHD on a mg/m2 basis) from embryonic implantation through lactation had no effect on pup growth or development.
8.2 Lactation
Risk Summary
Data from published literature report the presence of bupropion and its metabolites in human milk. Limited data from postmarketing reports with bupropion use during lactation have not identified a clear association of adverse effects on a breastfed infant (see Data). Naltrexone and its major metabolite, 6β-naltrexol, are present in human milk. There are no data on bupropion, naltrexone, or their metabolites on milk production. The developmental and health benefits of breastfeeding should be considered along with the mother's clinical need for CONTRAVE and any potential adverse effects on the breastfed infant from CONTRAVE or from the mother's underlying condition.
Data
In a lactation study of ten women, levels of orally dosed bupropion and its active metabolites were measured in expressed milk. The average daily infant exposure (assuming 150 mL/kg daily consumption) to bupropion and its active metabolites was 2% of the maternal weight-adjusted dose. Postmarketing reports have described seizures in breastfed infants. The relationship of bupropion exposure and these seizures is unclear.
8.4 Pediatric Use
The safety and effectiveness of CONTRAVE in pediatric patients below the age of 18 have not been established and the use of CONTRAVE is not recommended in pediatric patients.
8.5 Geriatric Use
Of the 3,239 subjects who participated in clinical trials with CONTRAVE, 62 (2%) were 65 years and older and none were 75 years and older. Clinical studies of CONTRAVE did not include sufficient numbers of subjects aged 65 and over to determine whether they respond differently from younger subjects. Older individuals may be more sensitive to the central nervous system adverse effects of CONTRAVE. Naltrexone and bupropion are known to be substantially excreted by the kidney, and the risk of adverse reactions to CONTRAVE may be greater in patients with impaired renal function. Because elderly patients are more likely to have decreased renal function, care should be taken in dose selection, and it may be useful to monitor renal function. CONTRAVE should be used with caution in patients over 65 years of age.
8.6 Renal Impairment
In a pharmacokinetic study conducted for CONTRAVE in subjects with renal impairment (mild, moderate and severe), exposure to naltrexone metabolite, 6-beta naltrexol, and bupropion metabolites, threohydrobupropion, and erythrohydrobupropion was increased. Therefore, the maximum recommended daily maintenance dose for CONTRAVE is two tablets (one tablet each morning and evening) in patients with moderate or severe renal impairment. CONTRAVE is not recommended for use in patients with end-stage renal disease [see Dosage and Administration (2.2) and Clinical Pharmacology (12.3)].
8.7 Hepatic Impairment
In a pharmacokinetic study conducted for CONTRAVE in subjects with hepatic impairment (mild, moderate, and severe), exposure to naltrexone, bupropion, and their metabolites were increased. Therefore, the maximum recommended daily maintenance dose of CONTRAVE is two tablets (one tablet each morning and evening) in patients with moderate hepatic impairment. CONTRAVE is not recommended for use in patients with severe hepatic impairment [see Dosage and Administration (2.3) and Clinical Pharmacology (12.3)].
- Pregnancy: Weight loss during pregnancy may cause fetal harm. Discontinue when a pregnancy is recognized. (8.1)
- Pediatric Use: Safety and effectiveness not established and use not recommended. (8.4)
CLINICAL PHARMACOLOGY SECTION
12 CLINICAL PHARMACOLOGY
12.1 Mechanism of Action
CONTRAVE has two components: naltrexone, an opioid antagonist, and bupropion, a relatively weak inhibitor of the neuronal reuptake of dopamine and norepinephrine. Nonclinical studies suggest that naltrexone and bupropion have effects on two separate areas of the brain involved in the regulation of food intake: the hypothalamus (appetite regulatory center) and the mesolimbic dopamine circuit (reward system). The exact neurochemical effects of CONTRAVE leading to weight loss are not fully understood.
12.2 Pharmacodynamics
Combined, bupropion and naltrexone increased the firing rate of hypothalamic pro-opiomelanocortin (POMC) neurons in vitro, which are associated with regulation of appetite. The combination of bupropion and naltrexone also reduced food intake when injected directly into the ventral tegmental area of the mesolimbic circuit in mice, an area associated with regulation of reward pathways.
Cardiac Electrophysiology
At the recommended dose, CONTRAVE does not prolong the QTc interval to any clinically relevant extent.
12.3 Pharmacokinetics
Absorption
Naltrexone
Following single oral administration of CONTRAVE (two 8 mg naltrexone/90 mg bupropion tablets) to healthy subjects, mean peak naltrexone concentration (Cmax) was 1.4 ng/mL, time to peak concentration (Tmax) was 2 hours, and extent of exposure (AUC0-inf) was 8.4 ng∙hr/mL.
Bupropion
Following single oral administration of CONTRAVE (two 8 mg naltrexone/90 mg bupropion tablets) to healthy subjects, mean peak bupropion concentration (Cmax) was 168 ng/mL, time to peak concentration (Tmax) was three hours, and extent of exposure (AUC0-inf) was 1,607 ng∙hr/mL.
Food Effect on Absorption
When CONTRAVE was administered with a high-fat meal, the AUC and Cmax for naltrexone increased 2.1-fold and 3.7-fold, respectively, and the AUC and Cmax for bupropion increased 1.4-fold and 1.8-fold, respectively. At steady state, the food effect increased AUC and Cmax for naltrexone by 1.7-fold and 1.9-fold, respectively, and increased AUC and Cmax for bupropion by 1.1-fold and 1.3-fold, respectively. Thus, CONTRAVE should not be taken with high-fat meals because of the resulting significant increases in bupropion and naltrexone systemic exposure.
Distribution
Naltrexone
Naltrexone is 21% plasma protein bound. The mean apparent volume of distribution at steady state for naltrexone (Vss/F) is 5,697 liters.
Bupropion
Bupropion is 84% plasma protein bound. The mean apparent volume of distribution at steady state for bupropion (Vss/F) is 880 liters.
Metabolism and Excretion
Naltrexone
The major metabolite of naltrexone is 6-beta-naltrexol. The activity of naltrexone is believed to be the result of both the parent and the 6-beta- naltrexol metabolite. Though less potent, 6-beta-naltrexol is eliminated more slowly and thus circulates at much higher concentrations than naltrexone. Naltrexone and 6-beta-naltrexol are not metabolized by cytochrome P450 enzymes and in vitro studies indicate that there is no potential for inhibition or induction of important isozymes.
Naltrexone and its metabolites are excreted primarily by the kidney (53% to 79% of the dose). Urinary excretion of unchanged naltrexone accounts for less than 2% of an oral dose. Urinary excretion of unchanged and conjugated 6-beta- naltrexol accounts for 43% of an oral dose. The renal clearance for naltrexone ranges from 30 to 127 mL/min, suggesting that renal elimination is primarily by glomerular filtration. The renal clearance for 6-beta-naltrexol ranges from 230 to 369 mL/min suggesting an additional renal tubular secretory mechanism. Fecal excretion is a minor elimination pathway.
Following single oral administration of CONTRAVE tablets to healthy subjects, mean elimination half-life (T1/2) was approximately 5 hours for naltrexone. Following twice daily administration of CONTRAVE, naltrexone did not accumulate and its kinetics appeared linear. However, in comparison to naltrexone, 6-beta-naltrexol accumulates to a larger extent (accumulation ratio ~3).
Bupropion
Bupropion is extensively metabolized with three active metabolites: hydroxybupropion, threohydrobupropion and erythrohydrobupropion. The metabolites have longer elimination half-lives than bupropion and accumulate to a greater extent. Following bupropion administration, more than 90% of the exposure is a result of metabolites. In vitro findings suggest that CYP2B6 is the principal isozyme involved in the formation of hydroxybupropion whereas cytochrome P450 isozymes are not involved in the formation of the other active metabolites. Bupropion and its metabolites inhibit CYP2D6. Plasma protein binding of hydroxybupropion is similar to that of bupropion (84%) whereas the other two metabolites have approximately half the binding.
Following oral administration of 200 mg of 14C-bupropion in humans, 87% and 10% of the radioactive dose were recovered in the urine and feces, respectively. The fraction of the oral dose of bupropion excreted unchanged was 0.5%, a finding consistent with the extensive metabolism of bupropion.
Following single oral administration of CONTRAVE tablets to healthy subjects, mean elimination half-life (T½) was approximately 21 hours for bupropion. Following twice daily administration of CONTRAVE, metabolites of bupropion, and to a lesser extent unchanged bupropion, accumulate and reach steady-state concentrations in approximately one week.
Specific Populations
Gender
Pooled analysis of CONTRAVE data suggested no clinically meaningful differences in the pharmacokinetic parameters of bupropion or naltrexone based on gender.
Race
Pooled analysis of CONTRAVE data suggested no clinically meaningful differences in the pharmacokinetic parameters of bupropion or naltrexone based on race.
Elderly
The pharmacokinetics of CONTRAVE have not been evaluated in the geriatric population. The effects of age on the pharmacokinetics of naltrexone or bupropion and their metabolites have not been fully characterized. An exploration of steady-state bupropion concentrations from several depression efficacy studies involving patients dosed in a range of 300 to 750 mg/day, on a three times daily schedule, revealed no relationship between age (18 to 83 years) and plasma concentration of bupropion. A single-dose pharmacokinetic study demonstrated that the disposition of bupropion and its metabolites in elderly subjects was similar to that of younger subjects. These data suggest there is no prominent effect of age on bupropion concentration; however, another pharmacokinetic study, single and multiple dose, has suggested that the elderly are at increased risk for accumulation of bupropion and its metabolites [see Use in Specific Populations (8.5)].
Smokers
Pooled analysis of CONTRAVE data revealed no meaningful differences in the plasma concentrations of bupropion or naltrexone in smokers compared with nonsmokers. The effects of cigarette smoking on the pharmacokinetics of bupropion were studied in 34 healthy male and female volunteers; 17 were chronic cigarette smokers and 17 were nonsmokers. Following oral administration of a single 150 mg dose of bupropion, there was no statistically significant difference in Cmax, half-life, Tmax, AUC, or clearance of bupropion or its active metabolites between smokers and nonsmokers.
Hepatic Impairment
A single dose pharmacokinetic study conducted for CONTRAVE, comparing patients with mild (n=8), moderate (n=8) and severe (n=7) hepatic impairment based on CHILD-PUGH classification system to subjects with normal hepatic function (n=13), showed that hepatic impairment had a significant effect on the PK parameters of the parent drugs naltrexone and bupropion. Systemic exposure to some metabolites was also increased in patients with impaired hepatic function [see Dosage and Administration (2.3) and Use in Specific Populations (8.7)].
Following a single dose of naltrexone/bupropion, AUCinf of naltrexone was approximately 2.8-, 6.1-, and 34-fold higher in patients with mild, moderate, and severe hepatic impairment, respectively. In patients with moderate and severe hepatic impairment, AUCinf of bupropion was approximately 2.0- and 3.6-fold higher, respectively, compared to subjects with normal hepatic function. There was no effect of mild hepatic impairment on bupropion exposure. Exposure to bupropion metabolite, threohydrobupropion, was increased by 1.9-, 3.4-, and 2.5-fold in patients with mild, moderate, and severe hepatic impairment, respectively [see Dosage and Administration (2.3) and Use in Specific Populations (8.7)].
Renal Impairment
A single-dose pharmacokinetic study conducted for CONTRAVE, comparing patients with mild (n=8), moderate (n=8) and severe (n=7) renal impairment to subjects with normal renal function (n=13), showed that renal impairment had no significant effect on the PK parameters of the parent drugs naltrexone and bupropion. However, systemic exposure (AUCinf) of some metabolites was increased in patients with impairment of renal function [see Dosage and Administration (2.2) and Use in Specific Populations (8.6)].
Following a single-dose of 16 mg naltrexone /180 mg bupropion, AUCinf of 6-beta-naltrexol was approximately1.5-, 1.7-, and 2.2-fold higher in patients with mild, moderate, and severe renal impairment, respectively. In patients with mild, moderate, and severe renal impairment, AUC of bupropion metabolites threohydrobupropion and erythrohydrobupropion increased approximately 1.3-, 1.9-, and 1.7-fold and 1.2-, 1.8-, and 1.5-fold, respectively. No studies have been conducted for CONTRAVE in patients with end stage renal disease.
The following information is available for individual components.
In a study of seven patients with end-stage renal disease requiring dialysis, peak plasma concentrations of naltrexone were elevated at least 6-fold compared to healthy subjects. An inter-trial comparison between normal subjects and patients with end-stage renal failure demonstrated that the bupropion Cmax and AUC values were comparable in the two groups, whereas the hydroxybupropion and threohydrobupropion metabolites had a 2.3- and 2.8-fold increase, respectively, in AUC for patients with end-stage renal failure.
Drug Interactions
In Vitro Assessment of Drug Interactions
At therapeutically relevant concentrations, naltrexone and 6-beta-naltrexol are not major inhibitors of CYP isoforms CYP1A2, CYP2B6, CYP2C8, CYP2E1, CYP2C9, CYP2C19, CYP2D6 or CYP3A4. Both naltrexone and 6-beta-naltrexol are not major inducers of CYP isoforms CYP1A2, CYP2B6, or CYP3A4.
Bupropion and its metabolites (hydroxybupropion, erythrohydrobupropion, threohydrobupropion) are inhibitors of CYP2D6.
In vitro studies suggest that paroxetine, sertraline, norfluoxetine, fluvoxamine, and nelfinavir inhibit the hydroxylation of bupropion.
Bupropion (IC50 9.3 mcM) and its metabolites, hydroxybupropion (IC50 82 mcM) and threohydrobupropion and erythrohydrobupropion (1:1 mixture; IC50 7.8 mcM), inhibited the renal organic transporter OCT2 to a clinically relevant level.
Effects of Naltrexone/Bupropion on the Pharmacokinetics of Other Drugs
Drug interaction between CONTRAVE and CYP2D6 substrates (metoprolol) or other drugs (atorvastatin, glyburide, lisinopril, nifedipine, valsartan) has been evaluated. In addition, drug interaction between bupropion, a component of CONTRAVE, and CYP2D6 substrates (desipramine) or other drugs (citalopram, lamotrigine) has also been evaluated.
Table 4. Effect of Naltrexone/Bupropion Coadministration on Systemic Exposure of Other Drugs|
Naltrexone/Bupropion Dosage |
Coadministered Drug | |
|---|---|---|
|
Name and Dose Regimens |
Change in Systemic Exposure | |
|
Initiate the following drugs at the lower end of the dose range during concomitant use with CONTRAVE[see Drug Interactions 7]: | ||
|
Bupropion |
Desipramine |
↑5-fold AUC, ↑2-fold Cmax |
|
Bupropion |
Citalopram |
↑40% AUC, ↑30% Cmax |
|
Naltrexone/Bupropion |
Metoprolol |
↑4-fold AUC, ↑2-fold Cmax |
|
No dose adjustment needed for the following drugs during concomitant use with CONTRAVE: | ||
|
Naltrexone/Bupropion |
Atorvastatin |
No Effect |
|
Naltrexone/Bupropion |
Glyburide |
No Effect |
|
Naltrexone/Bupropion |
Lisinopril |
No Effect |
|
Naltrexone/Bupropion |
Metformin |
↑23% AUC; |
|
Naltrexone/Bupropion |
Nifedipine |
No Effect |
|
Naltrexone/Bupropion |
Valsartan |
No Effect |
|
Bupropion |
Lamotrigine |
No Effect |
Digoxin: Literature data showed that digoxin exposure was decreased when a single oral dose of 0.5 mg digoxin was administered 24 hours after a single oral dose of extended-release 150 mg bupropion in healthy volunteers.
Effects of Other Drugs on the Pharmacokinetics of Naltrexone/Bupropion
Drug interactions between CYP2B6 inhibitors (ticlopidine, clopidogrel, prasugrel), CYP2B6 inducers (ritonavir, lopinavir) and bupropion (one of the CONTRAVE components), or between other drugs (atorvastatin, glyburide, metoprolol, lisinopril, nifedipine, valsartan) and CONTRAVE have been evaluated. While not systematically studied, carbamazepine, phenobarbital, or phenytoin may induce the metabolism of bupropion.
Table 5. Effect of Coadministered Drugs on Systemic Exposure of Naltrexone/Bupropion|
Name and Dose Regimens |
Coadministered Drug | |
|---|---|---|
|
CONTRAVE Components |
Change in Systemic Exposure | |
| ||
|
Do not exceed one tablet twice daily dose of CONTRAVE with the following drugs: | ||
|
Ticlopidine |
Bupropion |
↑85% AUC, ↑38% Cmax |
|
Clopidogrel |
Bupropion |
↑60% AUC, ↑40% Cmax |
|
No dose adjustment needed for CONTRAVE with the following drugs: | ||
|
Atorvastatin |
Naltrexone |
No Effect |
|
Lisinopril |
Naltrexone |
No Effect |
|
Valsartan |
Naltrexone |
No Effect |
|
Cimetidine |
Bupropion |
No Effect |
|
Citalopram |
Bupropion |
No Effect |
|
Metoprolol |
Naltrexone |
↓25% AUC, ↓29% Cmax |
|
Metformin |
Bupropion |
No Effect |
|
Nifedipine |
Naltrexone |
↑24% AUC, ↑58% Cmax |
|
Prasugrel |
Bupropion |
↑18% AUC, ↑14% Cmax |
|
Use CONTRAVE with caution with the following drugs: | ||
|
Glyburide |
Naltrexone |
↑2-fold AUC, ↑2-fold Cmax |
|
Avoid concomitant use of CONTRAVE with following drugs: | ||
|
Ritonavir |
Bupropion |
↓22% AUC, ↓21 % Cmax |
|
600 mg twice daily for 8 days |
Bupropion |
↓66% AUC, ↓62% Cmax |
|
Lopinavir/Ritonavir |
Bupropion |
↓57% AUC, ↓57% Cmax |
|
Efavirenz |
Bupropion |
↓55% AUC, ↓34% Cmax |
CLINICAL STUDIES SECTION
14 CLINICAL STUDIES
The effects of CONTRAVE on weight loss in conjunction with reduced caloric intake and increased physical activity was studied in double-blind, placebo- controlled trials (BMI range 27 to 45 kg/m2) with study durations of 16 to 56 weeks randomized to naltrexone and/or bupropion or placebo.
Effect on Weight Loss and Weight Maintenance
Four 56-week multicenter, double-blind, placebo-controlled obesity trials (CONTRAVE Obesity Research, or COR-I, COR-II, COR-BMOD, and COR-Diabetes) were conducted to evaluate the effect of CONTRAVE in conjunction with lifestyle modification in 4,536 patients randomized to CONTRAVE or placebo. The COR-I, COR-II, and COR-BMOD trials enrolled patients with obesity (BMI 30 kg/m2 or greater) or overweight (BMI 27 kg/m2 or greater) and at least one comorbidity (hypertension or dyslipidemia). The COR-Diabetes trial enrolled patients with BMI greater than 27 kg/m2 with type 2 diabetes with or without hypertension and/or dyslipidemia.
Treatment was initiated with a three-week dose-escalation period followed by approximately 1 year of continued therapy. Patients were instructed to take CONTRAVE with food. COR-I and COR-II included a program consisting of a reduced-calorie diet resulting in an approximate 500 kcal/day decrease in caloric intake, behavioral counseling, and increased physical activity. COR- BMOD included an intensive behavioral modification program consisting of 28 group counseling sessions over 56 weeks as well as a prescribed diet and exercise regimen. COR-Diabetes evaluated patients with type 2 diabetes not achieving glycemic goal of a HbA1c less than 7% either with oral antidiabetic agents or with diet and exercise alone. Of the overall population from these four trials, 24% had hypertension, 54% had dyslipidemia at study entry, and 10% had type 2 diabetes.
Apart from COR-Diabetes, which only enrolled patients with type 2 diabetes, the demographic characteristics of patients were similar across all four trials. For the four trial populations combined, the mean age was 46 years, 83% were female, 77% were Caucasian, 18% were black, and 5% were other races. At baseline, mean BMI was 36 kg/m2 and mean waist circumference was 110 cm.
A substantial percentage of randomized patients withdrew from the trials prior to Week 56: 45% for the placebo group and 46% for the CONTRAVE group. The majority of these patients discontinued within the first 12 weeks of treatment. Approximately 24% of patients treated with CONTRAVE and 12% of patients treated with placebo discontinued treatment because of an adverse reaction [see Adverse Reactions (6.1)].
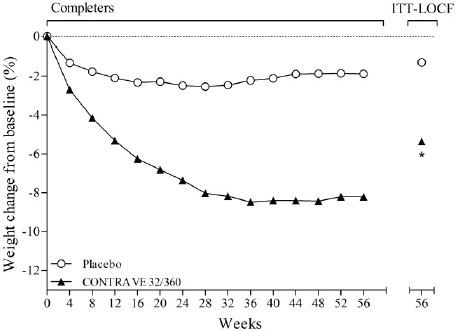
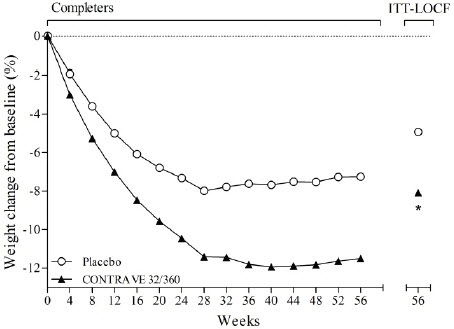
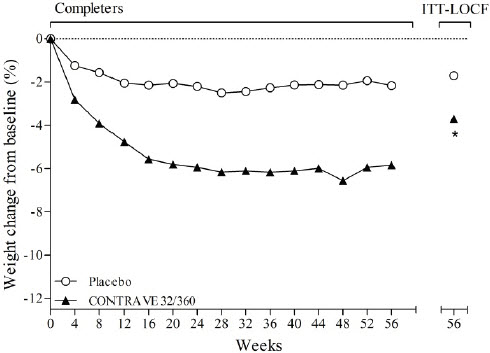
The co-primary endpoints were percent change from baseline body weight and the proportion of patients achieving at least a 5% reduction in body weight. In the 56-week COR-I trial, the mean change in body weight was -5.4% among patients assigned to CONTRAVE 32 mg/360 mg compared with -1.3% among patients assigned to placebo (Intent-To-Treat [ITT] population), as shown in Table 6 and Figure 1. In this trial, the achievement of at least a 5% reduction in body weight from baseline occurred more frequently for patients treated with CONTRAVE 32 mg/360 mg compared with placebo (42% vs 17%; Table 6). Results from COR-BMOD and COR-Diabetes are shown in Table 6 and Figures 2 and 3.
Table 6. Changes in Weight in 56-Week Trials with CONTRAVE (ITT/LOCF*)|
COR-I |
COR-BMOD |
COR-Diabetes | ||||
|---|---|---|---|---|---|---|
|
CONTRAVE |
Placebo |
CONTRAVE |
Placebo |
CONTRAVE |
Placebo | |
|
N |
538 |
536 |
565 |
196 |
321 |
166 |
|
Type 1 error was controlled across all 3 endpoints | ||||||
| ||||||
|
Weight (kg) | ||||||
|
Baseline mean (SD) |
99.8 |
99.5 |
100.3 |
101.8 |
104.2 |
105.3 |
|
LS Mean % Change From Baseline (SE) |
-5.4 |
-1.3 |
-8.1 |
-4.9 |
-3.7 |
-1.7 |
|
Difference from placebo (95% CI) |
-4.1† |
-3.2† |
-2.0† | |||
|
Percentage of patients losing greater than or equal to 5% body weight |
42 |
17 |
57 |
43 |
36 |
18 |
|
Risk difference vs placebo (95% CI) |
25† |
14† |
18† | |||
|
Percentage of patients losing greater than or equal to 10% body weight |
21 |
7 |
35 |
21 |
15 |
5 |
|
Risk difference vs placebo (95% CI) |
14† |
14† |
10‡ |
The percentages of patients who achieved at least 5% or at least 10% body weight loss from baseline were greater among those assigned to CONTRAVE, compared with placebo, in all four obesity trials (Table 6).
|
Figure 1. Weight Loss Over Time in Completer Population: COR-I Trial |
Figure 2. Weight Loss Over Time in Completer Population: COR-BMOD Trial |
|
|
|
|
*p<0.001 vs placebo |
*p<0.001 vs placebo COR-BMOD trial: 41.6% in the placebo group and 42.1% in the CONTRAVE group discontinued study drug. |
|
Figure 3. Weight Loss Over Time in Completer Population: COR-Diabetes Trial | ||
|
| ||
|
*p<0.001 vs placebo |
Effect on Cardiovascular and Metabolic Parameters
Changes in cardiovascular and metabolic parameters associated with obesity are presented for COR-I and COR-BMOD (Table 7). Changes in mean blood pressure and heart rate are further described elsewhere [see Warnings and Precautions (5.5)].
Table 7. Change in Markers of Cardiovascular and Metabolic Parameters from Baseline in 56 Week Trials with CONTRAVE 32 mg/360 mg (COR-I and COR- BMOD)*|
Parameter |
COR-I |
COR-BMOD | ||||
|---|---|---|---|---|---|---|
|
CONTRAVE |
Placebo |
CONTRAVE minus Placebo |
CONTRAVE |
Placebo |
CONTRAVE minus Placebo | |
|
Q1: first quartile; Q3: third quartile | ||||||
| ||||||
|
Triglycerides, mg/dL | ||||||
|
Baseline median (Q1, Q3) |
113 |
112 |
-10.7† |
110 |
103 |
-9.9† |
|
Median % change |
-11.6 |
1.7 |
-17.8 |
-7.4 | ||
|
HDL-C, mg/dL | ||||||
|
Baseline mean (SD) |
51.9 |
52.0 |
7.2 |
53.6 |
55.3 |
6.6 |
|
LS Mean % change (SE) |
8.0 |
0.8 |
9.4 |
2.8 | ||
|
LDL-C, mg/dL | ||||||
|
Baseline mean (SD) |
118.8 |
119.7 |
-1.5 |
109.5 |
109.2 |
-2.9 |
|
LS Mean % change (SE) |
-2.0 |
-0.5 |
7.1 |
10.0 | ||
|
Waist circumference, cm | ||||||
|
Baseline mean (SD) |
108.8 |
110.0 |
-3.8‡ |
109.3 |
109.0 |
-3.2‡ |
|
LS Mean change (SE) |
-6.2 |
-2.5 |
-10.0 |
-6.8 | ||
|
Heart rate, bpm | ||||||
|
Baseline mean (SD) |
72.1 |
71.8 |
1.2 |
70.7 |
70.4 |
0.9 |
|
LS Mean change (SE) |
1.0 |
-0.2 |
1.1 |
0.2 | ||
|
Systolic blood pressure, mmHg | ||||||
|
Baseline mean (SD) |
118.9 |
119.0 |
1.8 |
116.9 |
116.7 |
2.6 |
|
LS Mean change (SE) |
-0.1 |
-1.9 |
-1.3 |
-3.9 | ||
|
Diastolic blood pressure, mmHg | ||||||
|
Baseline mean (SD) |
77.1 |
77.3 |
0.9 |
78.2 |
77.2 |
1.4 |
|
LS Mean change (SE) |
0.0 |
-0.9 |
-1.4 |
-2.8 |
Effect of CONTRAVE on Cardiometabolic Parameters and Anthropometry in Patients with Type 2 Diabetes Mellitus
Changes in glycemic control observed from baseline to Week 56 among patients with type 2 diabetes and obesity assigned to either CONTRAVE 32 mg/360 mg or placebo are shown in Table 8.
Table 8. Changes in Cardiometabolic Parameters and Waist Circumference in Patients with Type 2 Diabetes Mellitus in a 56 Week Trial with CONTRAVE 32 mg/360 mg (COR-Diabetes)|
CONTRAVE |
Placebo | ||||
|---|---|---|---|---|---|
|
Based on last observation carried forward (LOCF) while on study drug | |||||
| |||||
|
Baseline |
Change from Baseline |
Baseline |
Change from Baseline |
CONTRAVE minus Placebo | |
|
HbA1c (%) |
8.0 |
-0.6 |
8.0 |
-0.1 |
-0.5* |
|
Fasting Glucose (mg/dL) |
160.0 |
-11.9 |
163.9 |
-4.0 |
-7.9 |
|
Waist Circumference (cm) |
115.6 |
-5.0 |
114.3 |
-2.9 |
-2.1 |
|
Systolic blood pressure (mmHg) |
125.0 |
0.0 |
124.5 |
-1.1 |
1.2 |
|
Diastolic blood pressure (mmHg) |
77.5 |
-1.1 |
77.4 |
-1.5 |
0.4 |
|
Heart rate (bpm) |
72.9 |
0.7 |
73.1 |
-0.2 |
0.9 |
|
Baseline |
% Change from Baseline |
Baseline |
% Change from Baseline |
CONTRAVE minus Placebo | |
|
Triglycerides (mg/dL)† |
147 (98, 200) |
-7.7 |
168 (114, 236) |
-8.6 |
-3.3 |
|
HDL Cholesterol (mg/dL) |
46.2 |
7.4 |
46.1 |
-0.2 |
7.6 |
|
LDL Cholesterol (mg/dL) |
100.2 |
2.4 |
101.0 |
4.2 |
-1.9 |
Effect on Body Composition
In a subset of 124 patients (79 CONTRAVE, 45 placebo), body composition was measured using dual energy X-ray absorptiometry (DEXA). The DEXA assessment showed that mean total body fat mass decreased by 4.7 kg (11.7%) in the CONTRAVE group vs 1.4 kg (4.3%) in the placebo group at Week 52/LOCF (treatment difference, -3.3 kg [-7.4%], p<0.01).