
COMPLERA Access
These highlights do not include all the information needed to use COMPLERA safely and effectively. See full prescribing information for COMPLERA. COMPLERA™ (emtricitabine, rilpivirine, tenofovir disoproxil fumarate) tablets, for oral use GILEAD ACCESS PROGRAM Initial U.S. Approval: 2011
9176bf31-c889-47bd-861c-f2d3fc37ab8d
HUMAN PRESCRIPTION DRUG LABEL
Feb 20, 2024
Gilead Sciences, Inc.
DUNS: 185049848
Products 1
Detailed information about drug products covered under this FDA approval, including NDC codes, dosage forms, ingredients, and administration routes.
EMTRICITABINE, RILPIVIRINE HYDROCHLORIDE, and TENOFOVIR DISOPROXIL FUMARATE
Product Details
FDA regulatory identification and product classification information
FDA Identifiers
Product Classification
Product Specifications
INGREDIENTS (14)
Drug Labeling Information
PACKAGE LABEL.PRINCIPAL DISPLAY PANEL
PRINCIPAL DISPLAY PANEL - 30 Tablet Bottle Label
61958-1102-5
30 tablets
COMPLERA™
(emtricitabine, rilpivirine,
tenofovir disoproxil fumarate) tablets
200 mg / 25 mg / 300 mg
GILEAD ACCESS PROGRAM
Note to pharmacist: Do not cover ALERT box with pharmacy label.
ALERT: Find out about medicines that
should NOT be taken with COMPLERA
Rx only
POM

BOXED WARNING SECTION
WARNING: POSTTREATMENT ACUTE EXACERBATION OF HEPATITIS B
CONTRAINDICATIONS SECTION
4 CONTRAINDICATIONS
COMPLERA is contraindicated when coadministered with the following drugs; coadministration may result in loss of virologic response and possible resistance to COMPLERA or to the class of NNRTIs [see Warnings and Precautions (5.7), Drug Interactions (7), and Clinical Pharmacology (12.3)]:
- Anticonvulsants: carbamazepine, oxcarbazepine, phenobarbital, phenytoin
- Antimycobacterials: rifampin, rifapentine
- Glucocorticoid (systemic): dexamethasone (more than a single-dose)
- Herbal Products: St John's wort (Hypericum perforatum)
- Proton Pump Inhibitors: e.g., dexlansoprazole, esomeprazole, lansoprazole, omeprazole, pantoprazole, rabeprazole
COMPLERA is contraindicated when coadministered with drugs which may result in loss of virologic response and possible resistance to COMPLERA. (4)
ADVERSE REACTIONS SECTION
6 ADVERSE REACTIONS
The following adverse reactions are discussed in other sections of the labeling:
- Severe Acute Exacerbations of Hepatitis B in Patients Coinfected with HIV-1 and HBV [see Warnings and Precautions (5.1)].
- Skin and Hypersensitivity Reactions [see Warnings and Precautions (5.2)].
- Hepatotoxicity [see Warnings and Precautions (5.3)].
- Depressive Disorders [see Warnings and Precautions (5.4)].
- New Onset or Worsening Renal Impairment [see Warnings and Precautions (5.5)].
- Bone Loss and Mineralization Defects [see Warnings and Precautions (5.6)].
- Lactic Acidosis/Severe Hepatomegaly with Steatosis [see Warnings and Precautions (5.8)].
- Immune Reconstitution Syndrome [see Warnings and Precautions (5.9)].
6.1 Clinical Trials Experience
Because clinical trials are conducted under widely varying conditions, adverse reaction rates observed in the clinical trials of a drug cannot be directly compared to rates in the clinical trials of another drug and may not reflect the rates observed in practice.
Adverse Reactions from Clinical Trials Experience in Adult Subjects
In HIV-1-Infected Adult Subjects With No Antiretroviral Treatment History
Studies C209 and C215
The safety assessment of RPV, used in combination with other antiretroviral drugs, is based on the Week 96 pooled data from 1368 subjects in the Phase 3 trials TMC278-C209 (ECHO) and TMC278-C215 (THRIVE) in antiretroviral treatment-naïve HIV-1-infected adult subjects. A total of 686 subjects received RPV in combination with other antiretroviral drugs as background regimen; most (N=550) received FTC/TDF as background regimen. The number of subjects randomized to the control arm EFV was 682, of which 546 received FTC/TDF as background regimen [see Clinical Studies (14)]. The median duration of exposure for subjects in either treatment arm was 104 weeks.
Adverse reactions observed at Week 96 in subjects who received RPV or EFV + FTC/TDF as background regimen are shown in Table 1. No new types of adverse reactions were identified between Week 48 and Week 96. The adverse reactions observed in this subset of subjects were generally consistent with those seen for the overall patient population participating in these studies (refer to the prescribing information for Edurant).
The proportion of subjects who discontinued treatment with RPV or EFV + FTC/TDF due to adverse reactions, regardless of severity, was 2% and 5%, respectively. The most common adverse reactions leading to discontinuation were psychiatric disorders: 9 (1.6%) subjects in the RPV + FTC/TDF arm and 12 (2.2%) subjects in the EFV + FTC/TDF arm. Rash led to discontinuation in 1 (0.2%) subject in the RPV + FTC/TDF arm and 10 (1.8%) subjects in the EFV + FTC/TDF arm.
Common Adverse Reactions: Clinical adverse reactions to RPV or EFV of at least moderate intensity (≥Grade 2) reported in at least 2% of adult subjects are shown in Table 1.
Table 1 Selected Adverse Reactions* (Grades 2–4) Reported in ≥2% of Adult Subjects Receiving RPV or EFV in Combination with FTC/TDF in Studies C209 and C215 (Week 96 Analysis)|
Preferred Term |
RPV + FTC/TDF |
EFV + FTC/TDF |
|---|---|---|
|
N=550 |
N=546 | |
| ||
|
Depressive disorders† |
2% |
2% |
|
Headache |
2% |
2% |
|
Insomnia |
2% |
2% |
|
Abnormal dreams |
1% |
3% |
|
Dizziness |
1% |
7% |
|
Nausea |
1% |
2% |
|
Rash |
1% |
5% |
Rilpivirine: Adverse reactions of at least moderate intensity (≥Grade 2) that occurred in less than 2% of subjects treated with RPV plus any of the allowed background regimens (N=686) in clinical studies C209 and C215 include (grouped by Body System): vomiting, diarrhea, abdominal discomfort, abdominal pain, fatigue, cholecystitis, cholelithiasis, decreased appetite, somnolence, sleep disorders, anxiety, glomerulonephritis membranous, glomerulonephritis mesangioproliferative, and nephrolithiasis.
In Virologically Suppressed HIV-1-Infected Adult Subjects
No new adverse reactions to COMPLERA were identified in stable, virologically suppressed subjects switching to COMPLERA from a regimen containing a ritonavir-boosted protease inhibitor; however, the frequency of adverse reactions increased by 20% (Study 106) after switching to COMPLERA.
Emtricitabine and Tenofovir DF: The most common adverse reactions that occurred in at least 10% of HIV-1-infected treatment-naïve adult subjects in a Phase 3 clinical trial of FTC and TDF in combination with another antiretroviral agent were diarrhea, nausea, fatigue, headache, dizziness, depression, insomnia, abnormal dreams, and rash. Adverse reactions that occurred in at least 5% of treatment-experienced or treatment-naïve subjects receiving FTC or TDF with other antiretroviral agents in clinical trials included abdominal pain, dyspepsia, vomiting, fever, pain, nasopharyngitis, pneumonia, sinusitis, upper respiratory tract infection, arthralgia, back pain, myalgia, paresthesia, peripheral neuropathy (including peripheral neuritis and neuropathy), anxiety, increased cough, and rhinitis.
Skin discoloration has been reported with higher frequency among FTC-treated subjects; it was manifested by hyperpigmentation on the palms and/or soles and was generally mild and asymptomatic. The mechanism and clinical significance are unknown.
Laboratory Abnormalities in Adult Subjects
The percentage of subjects treated with RPV + FTC/TDF or EFV + FTC/TDF in studies C209 and C215 with selected laboratory abnormalities (Grades 1–4), representing worst-grade toxicity, is presented in Table 2.
Table 2 Selected Laboratory Abnormalities (Grades 1–4) Reported in Adult Subjects Who Received RPV or EFV in Combination with FTC/TDF in Studies C209 and C215 (Week 96 Analysis)|
Laboratory Parameter Abnormality |
DAIDS Toxicity Range |
RPV + FTC/TDF |
EFV + FTC/TDF |
|---|---|---|---|
|
N=550 |
N=546 | ||
|
N=number of subjects per treatment group | |||
|
BIOCHEMISTRY | |||
|
Increased Creatinine | |||
|
Grade 1 |
1.1–1.3 × ULN |
6% |
1% |
|
Grade 2 |
|
1% |
1% |
|
Grade 3 |
|
<1% |
0 |
|
Grade 4 |
|
0 |
<1% |
|
Increased AST | |||
|
Grade 1 |
1.25–2.5 × ULN |
16% |
19% |
|
Grade 2 |
|
4% |
7% |
|
Grade 3 |
|
2% |
3% |
|
Grade 4 |
|
1% |
1% |
|
Increased ALT | |||
|
Grade 1 |
1.25–2.5 × ULN |
19% |
22% |
|
Grade 2 |
|
5% |
7% |
|
Grade 3 |
|
1% |
2% |
|
Grade 4 |
|
1% |
1% |
|
Increased Total Bilirubin | |||
|
Grade 1 |
1.1–1.5 × ULN |
6% |
<1% |
|
Grade 2 |
|
3% |
1% |
|
Grade 3 |
|
1% |
<1% |
|
Increased Total Cholesterol (fasted) | |||
|
Grade 1 |
200–239 mg/dL |
14% |
31% |
|
Grade 2 |
240–300 mg/dL |
6% |
18% |
|
Grade 3 |
|
<1% |
2% |
|
Increased LDL Cholesterol (fasted) | |||
|
Grade 1 |
130–159 mg/dL |
13% |
28% |
|
Grade 2 |
160–190 mg/dL |
5% |
13% |
|
Grade 3 |
|
1% |
4% |
|
Increased Triglycerides (fasted) | |||
|
Grade 2 |
500–750 mg/dL |
1% |
2% |
|
Grade 3 |
751–1200 mg/dL |
1% |
2% |
|
Grade 4 |
|
0 |
1% |
Emtricitabine or Tenofovir DF: The following Grade 3 or 4 laboratory abnormalities have been previously reported in subjects treated with FTC or TDF with other antiretroviral agents in other clinical trials: increased pancreatic amylase (>2.0 × ULN), increased serum amylase (>175 U/L), increased lipase (>3.0 × ULN), increased alkaline phosphatase (>550 U/L), increased or decreased serum glucose (<40 or >250 mg/dL), increased glycosuria (≥3+), increased creatine kinase (M: >990 U/L; F: >845 U/L), decreased neutrophils (<750/mm3), and increased hematuria (>75 RBC/HPF).
Adrenal Function: In the pooled Phase 3 trials of C209 and C215, in subjects treated with RPV plus any of the allowed background regimens (N=686), at Week 96 there was an overall mean change from baseline in basal cortisol of –0.69 (–1.12, 0.27) micrograms/dL in the RPV group, and of –0.02 (–0.48, 0.44) micrograms/dL in the EFV group.
In the RPV group, 43/588 (7.3%) of subjects with a normal 250 micrograms ACTH stimulation test at baseline developed an abnormal 250 micrograms ACTH stimulation test (peak cortisol level <18.1 micrograms/dL) during the trial compared to 18/561 (3.2%) in the EFV group. Of the subjects who developed an abnormal 250 micrograms ACTH stimulation test during the trial, 14 subjects in the RPV group and 9 subjects in the EFV group had an abnormal 250 micrograms ACTH stimulation test at Week 96. Overall, there were no serious adverse events, deaths, or treatment discontinuations that could clearly be attributed to adrenal insufficiency. The clinical significance of the higher abnormal rate of 250 micrograms ACTH stimulation tests in the RPV group is not known.
Serum Creatinine: In the pooled Phase 3 trials of C209 and C215 in subjects treated with RPV plus any of the allowed background regimens (N=686), there was a small increase in serum creatinine over 96 weeks of treatment with RPV. Most of this increase occurred within the first 4 weeks of treatment, with a mean change of 0.1 mg/dL (range –0.3 to 0.6 mg/dL) observed through Week 96. In subjects who entered the trial with mild or moderate renal impairment, the serum creatinine increase observed was similar to that seen in subjects with normal renal function. These changes are not considered to be clinically relevant, and no subject discontinued treatment due to increases in serum creatinine. Creatinine increases were comparable by background N(t)RTIs.
Serum Lipids: Changes from baseline in total cholesterol, LDL-cholesterol, and triglycerides are presented in Table 3.
Table 3 Lipid Values Reported in Adult Subjects Receiving RPV or EFV in Combination with FTC/TDF in Studies C209 and C215*|
Pooled Data from the Week 96 Analysis of C209 and C215 Trials | ||||||||
|---|---|---|---|---|---|---|---|---|
|
RPV + FTC/TDF |
EFV + FTC/TDF | |||||||
|
N |
Baseline |
Week 96 |
N |
Baseline |
Week 96 | |||
|
Mean |
Mean |
Mean |
Mean Change† |
Mean |
Mean |
Mean Change† | ||
|
N=number of subjects per treatment group | ||||||||
| ||||||||
|
Total Cholesterol (fasted) |
430 |
162 |
164 |
2 |
401 |
160 |
186 |
26 |
|
HDL-cholesterol (fasted) |
429 |
42 |
45 |
4 |
399 |
40 |
50 |
11 |
|
LDL-cholesterol (fasted) |
427 |
97 |
97 |
–1 |
397 |
96 |
110 |
14 |
|
Triglycerides (fasted) |
430 |
123 |
109 |
–14 |
401 |
127 |
133 |
6 |
Adult Subjects Coinfected with Hepatitis B and/or Hepatitis C Virus: In adult subjects coinfected with hepatitis B or C virus receiving RPV in studies C209 and C215, the incidence of hepatic enzyme elevation was higher than in subjects receiving RPV who were not coinfected. The same increase was also observed in the EFV arm. The pharmacokinetic exposure of RPV in coinfected subjects was comparable to that in subjects without coinfection.
Adverse Reactions from Clinical Trials Experience in Pediatric Subjects
Emtricitabine: In addition to the adverse reactions reported in adults, anemia and hyperpigmentation were observed in 7% and 32%, respectively, of pediatric subjects (3 months to less than 18 years of age) who received treatment with FTC in the larger of two open-label, uncontrolled pediatric trials (N=116). For additional information, please consult the EMTRIVA™ prescribing information.
Rilpivirine: The safety assessment is based on the Week 48 analysis of the single-arm, open-label Phase 2 trial, TMC278-C213, in which 36 antiretroviral treatment-naïve HIV-1-infected subjects 12 to less than 18 years of age and weighing at least 32 kg received RPV (25 mg once daily) in combination with other antiretroviral agents. The median duration of exposure for subjects was 63.5 weeks. No subjects discontinued treatment due to adverse reactions. No new adverse reactions were identified compared to those seen in adults.
Adverse reactions were reported in 19 pediatric subjects (52.8%). Most adverse reactions were Grade 1 or 2. The most common adverse reactions reported in at least 2 subjects (regardless of severity) include headache (19.4%), depression (19.4%), somnolence (13.9%), nausea (11.1%), dizziness (8.3%), abdominal pain (8.3%), vomiting (5.6%), and rash (5.6%).
Observed laboratory abnormalities were comparable to those in adults. For additional information, please consult the Edurant prescribing information.
Adrenal Function
In trial TMC278-C213, at Week 48, the overall mean change from baseline in basal cortisol showed an increase of 1.59 (0.24, 2.93) micrograms/dL.
Six of 30 (20%) subjects with a normal 250 micrograms ACTH stimulation test at baseline developed an abnormal 250 micrograms ACTH stimulation test (peak cortisol level <18.1 micrograms/dL) during the trial. Three of these subjects had an abnormal 250 micrograms ACTH stimulation test at Week 48. Overall, there were no serious adverse events, deaths, or treatment discontinuations that could clearly be attributed to adrenal insufficiency. The clinical significance of the abnormal 250 micrograms ACTH stimulation tests is not known.
Tenofovir DF: In a pediatric clinical trial conducted in subjects 12 to less than 18 years of age, the adverse reactions observed in pediatric subjects who received treatment with TDF were consistent with those observed in clinical trials of TDF in adults [see Warnings and Precautions (5.6)]. For additional information, including information on bone mineral density changes, please consult the VIREAD™ prescribing information.
6.2 Postmarketing Experience
The following adverse reactions have been identified during postmarketing experience in patients receiving RPV- or TDF-containing regimens. Because postmarketing reactions are reported voluntarily from a population of uncertain size, it is not always possible to reliably estimate their frequency or establish a causal relationship to drug exposure.
COMPLERA:
Metabolism and Nutrition Disorders
weight increased
Skin and Subcutaneous Tissue Disorders
severe skin and hypersensitivity reactions including DRESS (Drug Reaction with Eosinophilia and Systemic Symptoms)
Rilpivirine:
Renal and Urinary Disorders
nephrotic syndrome
Emtricitabine:
No postmarketing adverse reactions have been identified for inclusion in this section.
Tenofovir DF:
Immune System Disorders
allergic reaction, including angioedema
Metabolism and Nutrition Disorders
lactic acidosis, hypokalemia, hypophosphatemia
Respiratory, Thoracic, and Mediastinal Disorders
dyspnea
Gastrointestinal Disorders
pancreatitis, increased amylase, abdominal pain
Hepatobiliary Disorders
hepatic steatosis, hepatitis, increased liver enzymes (most commonly AST, ALT, gamma GT)
Skin and Subcutaneous Tissue Disorders
rash
Musculoskeletal and Connective Tissue Disorders
rhabdomyolysis, osteomalacia (manifested as bone pain and which may contribute to fractures), muscular weakness, myopathy
Renal and Urinary Disorders
acute renal failure, renal failure, acute tubular necrosis, Fanconi syndrome, proximal renal tubulopathy, interstitial nephritis (including acute cases), nephrogenic diabetes insipidus, renal insufficiency, increased creatinine, proteinuria, polyuria
General Disorders and Administration Site Conditions
asthenia
The following adverse reactions, listed under the body system headings above, may occur as a consequence of proximal renal tubulopathy: rhabdomyolysis, osteomalacia, hypokalemia, muscular weakness, myopathy, hypophosphatemia.
- Most common adverse reactions to rilpivirine (incidence greater than or equal to 2%, Grades 2–4) are depressive disorders, insomnia, and headache. (6.1)
- Most common adverse reactions to emtricitabine and tenofovir disoproxil fumarate (incidence greater than or equal to 10%) are diarrhea, nausea, fatigue, headache, dizziness, depression, insomnia, abnormal dreams, and rash. (6.1)
To report SUSPECTED ADVERSE REACTIONS, contact Gilead Sciences, Inc. at safety_fc@gilead.com or the US FDA at 1-800-FDA-1088 or at www.fda.gov/medwatch
DRUG INTERACTIONS SECTION
7 DRUG INTERACTIONS
7.1 Not Recommended with Other Antiretroviral Medications
Because COMPLERA is a complete regimen, coadministration with other antiretroviral medications for the treatment of HIV-1 infection is not recommended. Comprehensive information regarding potential drug-drug interactions with other antiretroviral medications is not provided.
This section describes clinically relevant drug interactions with COMPLERA. Drug interaction studies were conducted with the components of COMPLERA (FTC, RPV, and TDF as single agents) or with COMPLERA as a combination product [see Dosage and Administration (2), Contraindications (4), and Clinical Pharmacology (12.3)].
7.2 Drugs Inducing or Inhibiting CYP3A Enzymes
Rilpivirine is primarily metabolized by cytochrome P450 (CYP) 3A, and drugs that induce or inhibit CYP3A may thus affect the clearance of RPV [see Contraindications (4), Warnings and Precautions (5.7), and Clinical Pharmacology (12.3)]. Coadministration of RPV and drugs that induce CYP3A may result in decreased plasma concentrations of RPV and loss of virologic response and possible resistance to RPV or to the class of NNRTIs. Coadministration of RPV and drugs that inhibit CYP3A may result in increased plasma concentrations of RPV.
7.3 Drugs Increasing Gastric pH
Coadministration of RPV with drugs that increase gastric pH may decrease plasma concentrations of RPV and loss of virologic response and possible resistance to RPV or to the class of NNRTIs. Use of RPV with proton pump inhibitors is contraindicated and use of RPV with H2-receptor antagonists requires staggered administration [see Contraindications (4) and Clinical Pharmacology (12.3)].
7.4 Drugs Affecting Renal Function
Because FTC and tenofovir are primarily eliminated by the kidneys through a combination of glomerular filtration and active tubular secretion, coadministration of COMPLERA with drugs that reduce renal function or compete for active tubular secretion may increase serum concentrations of FTC, tenofovir, and/or other renally eliminated drugs. Some examples of drugs that are eliminated by active tubular secretion include, but are not limited to, acyclovir, adefovir dipivoxil, cidofovir, ganciclovir, valacyclovir, valganciclovir, aminoglycosides (e.g., gentamicin), and high-dose or multiple NSAIDs [see Warnings and Precautions (5.5)].
7.5 QT Prolonging Drugs
There is limited information available on the potential for a pharmacodynamic interaction between RPV and drugs that prolong the QTc interval of the electrocardiogram. In a study of healthy subjects, 75 mg once daily and 300 mg once daily doses of RPV (3 times and 12 times the dose in COMPLERA) have been shown to prolong the QTc interval of the electrocardiogram [see Warnings and Precautions (5.7) and Clinical Pharmacology (12.2)]. Consider alternatives to COMPLERA when coadministered with a drug with a known risk of Torsade de Pointes.
7.6 Significant Drug Interactions
Important drug interaction information for COMPLERA is summarized in Table 4. The drug interactions described are based on studies conducted with FTC, RPV, or TDF as individual medications or with COMPLERA as a combination product, or are potential drug interactions [see Clinical Pharmacology (12.3), Tables 9–14]. For list of contraindicated drugs, [see Contraindications (4)].
Table 4 Significant* Drug Interactions|
Concomitant Drug Class: Drug Name |
Effect on Concentration† |
Clinical Comment |
|---|---|---|
| ||
|
Antacids: |
↔ RPV |
Administer antacids at least 2 hours before or at least 4 hours after COMPLERA. |
|
Anticonvulsants: |
↓ RPV |
Coadministration is contraindicated due to potential for loss of virologic response and development of resistance. |
|
Antimycobacterials: |
↓ RPV |
Coadministration is contraindicated due to potential for loss of virologic response and development of resistance. |
|
rifabutin |
↓ RPV‡ |
If COMPLERA is coadministered with rifabutin, an additional 25 mg tablet of RPV (Edurant) once per day is recommended to be taken concomitantly with COMPLERA and with a meal for the duration of rifabutin coadministration. |
|
Azole Antifungal Agents: |
↑ RPV‡,§ |
No dose adjustment is required when COMPLERA is coadministered with azole antifungal agents. Clinically monitor for breakthrough fungal infections when azole antifungals are coadministered with COMPLERA. |
|
Glucocorticoid (systemic): |
↓ RPV |
Coadministration is contraindicated due to potential for loss of virologic response and development of resistance. |
|
Hepatitis C Antiviral Agents: |
↑ tenofovir‡ |
Patients receiving COMPLERA concomitantly with HARVONI™ (ledipasvir/sofosbuvir), EPCLUSA™ (sofosbuvir/velpatasvir), or VOSEVI™ (sofosbuvir/velpatasvir/voxilaprevir) should be monitored for adverse reactions associated with TDF. |
|
H2-Receptor Antagonists: |
↔ RPV‡,§ |
Administer H2-receptor antagonists at least 12 hours before or at least 4 hours after COMPLERA. |
|
Herbal Products: |
↓ RPV |
Coadministration is contraindicated due to potential for loss of virologic response and development of resistance. |
|
Macrolide or Ketolide Antibiotics: |
↑ RPV |
Where possible, alternatives such as azithromycin should be considered. |
|
Narcotic Analgesics: |
↓ R(−) methadone‡ |
No dose adjustments are required when initiating coadministration of methadone with COMPLERA. However, clinical monitoring is recommended as methadone maintenance therapy may need to be adjusted in some patients. |
|
Proton Pump Inhibitors: |
↓ RPV |
Coadministration is contraindicated due to potential for loss of virologic response and development of resistance. |
7.7 Drugs with No Observed Interactions with COMPLERA
No clinically significant drug interactions have been observed between FTC and the following medications: famciclovir, ledipasvir/sofosbuvir, sofosbuvir/velpatasvir, sofosbuvir/velpatasvir/voxilaprevir, or TDF.
No clinically significant drug interactions have been observed between TDF and the following medications: entecavir, methadone, oral contraceptives, ribavirin, sofosbuvir, or tacrolimus in studies conducted in healthy subjects.
No clinically significant drug interactions have been observed between RPV and the following medications: acetaminophen, atorvastatin, chlorzoxazone, ethinyl estradiol, ledipasvir/sofosbuvir, norethindrone, sildenafil, simeprevir, sofosbuvir, sofosbuvir/velpatasvir, sofosbuvir/velpatasvir/voxilaprevir, or TDF. RPV did not have a clinically significant effect on the pharmacokinetics of digoxin or metformin.
- COMPLERA is a complete regimen for the treatment of HIV-1 infection; therefore, coadministration with other antiretroviral medications for treatment of HIV-1 infection is not recommended. (7.1)
- Consult the Full Prescribing Information prior to and during treatment for important drug interactions. (4, 5.7, 7)
RECENT MAJOR CHANGES SECTION
RECENT MAJOR CHANGES
|
Warnings and Precautions | |
|
Immune Reconstituition Syndrome (5.9) |
11/2019 |
DOSAGE FORMS & STRENGTHS SECTION
3 DOSAGE FORMS AND STRENGTHS
Each COMPLERA tablet contains 200 mg of emtricitabine (FTC), 27.5 mg of rilpivirine hydrochloride (equivalent to 25 mg of rilpivirine [RPV]), and 300 mg of tenofovir disoproxil fumarate (TDF, equivalent to 245 mg of tenofovir disoproxil).
The tablets are white, capsule shaped, film coated and debossed with "GSI" on both sides.
Tablets: 200 mg of emtricitabine, 25 mg of rilpivirine, and 300 mg of tenofovir disoproxil fumarate. (3)
SPL UNCLASSIFIED SECTION
Manufactured and distributed for:
Gilead Sciences, Inc.
Foster City, CA 94404
COMPLERA, EMTRIVA, EPCLUSA, GSI, HARVONI, ODEFSEY, TRUVADA, VIREAD, and VOSEVI are trademarks of Gilead Sciences, Inc., or its related companies. All other trademarks referenced herein are the property of their respective owners.
© 2019 Gilead Sciences, Inc. All rights reserved.
202123-GS-013A
SPL PATIENT PACKAGE INSERT SECTION
|
This Patient Information has been approved by the U.S. Food and Drug Administration. |
Revised: 11/2019 | |
|
Patient Information | ||
|
Important: Ask your healthcare provider or pharmacist about medicines that should not be taken with COMPLERA. For more information, see the section "What should I tell my healthcare provider before taking COMPLERA?" | ||
|
What is the most important information I should know about COMPLERA?
For more information about side effects, see the section "What are the possible side effects of COMPLERA?". | ||
|
What is COMPLERA?
HIV-1 is the virus that causes AIDS (Acquired Immune Deficiency Syndrome).
COMPLERA does not cure HIV-1 or AIDS. | ||
|
Who should not take COMPLERA?
| ||
|
What should I tell my healthcare provider before taking COMPLERA?
Tell your healthcare provider about all the medicines you take, including
prescription and over-the-counter medicines, vitamins, and herbal supplements.
| ||
|
How should I take COMPLERA?
| ||
|
What are the possible side effects of COMPLERA? *See "What is the most important information I should know about COMPLERA?" | ||
|
| |
|
*Severe liver problems. In rare cases, severe liver problems can happen that can lead to death. Tell your healthcare provider right away if you get these symptoms: skin or the white part of your eyes turns yellow, dark "tea-colored" urine, light-colored stools, loss of appetite for several days or longer, nausea, or stomach-area pain. *Change in liver enzymes. People with a history of hepatitis B or C virus infection or who have certain liver enzyme changes may have an increased risk of developing new or worsening liver problems during treatment with COMPLERA. Liver problems can also happen during treatment with COMPLERA in people without a history of liver disease. Your healthcare provider may need to do tests to check your liver enzymes before and during treatment with COMPLERA. *Depression or mood changes. Tell your healthcare provider right away if you have any of the following symptoms: * feel sad or hopeless * feel anxious or restless * have thoughts of hurting yourself (suicide) or have tried to hurt yourself *New or worse kidney problems, including kidney failure, can happen in some people who take COMPLERA. Your healthcare provider should do blood tests to check your kidneys before starting treatment with COMPLERA. If you have had kidney problems in the past or need to take another medicine that can cause kidney problems, your healthcare provider may need to do blood tests to check your kidneys during your treatment with COMPLERA. *Bone problems can happen in some people who take COMPLERA. Bone problems include bone pain, softening, or thinning (which may lead to fractures). Your healthcare provider may need to do additional tests to check your bones. *Too much lactic acid in your blood (lactic acidosis). Too much lactic acid is a serious but rare medical emergency that can lead to death. Tell your healthcare provider right away if you get these symptoms: weakness or being more tired than usual, unusual muscle pain, being short of breath or fast breathing, stomach pain with nausea and vomiting, cold or blue hands and feet, feel dizzy or lightheaded, or a fast or abnormal heartbeat. *Changes in your immune system (Immune Reconstitution Syndrome) can happen when you start taking HIV-1 medicines. Your immune system may get stronger and begin to fight infections that have been hidden in your body for a long time. Tell your healthcare provider right away if you start having new symptoms after starting your HIV-1 medicine. The most common side effects of rilpivirine, one of the medicines in COMPLERA, include:
The most common side effects of emtricitabine and tenofovir disoproxil fumarate, two of the medicines in COMPLERA, include: | ||
|
| |
|
These are not all the possible side effects of COMPLERA. | ||
|
How should I store COMPLERA?
Keep COMPLERA and all other medicines out of reach of children. | ||
|
General information about safe and effective use of COMPLERA | ||
|
What are the ingredients of COMPLERA? |
HOW SUPPLIED SECTION
16 HOW SUPPLIED/STORAGE AND HANDLING
COMPLERA tablets are white, capsule shaped, film coated, and debossed with "GSI" on both sides. Each bottle contains 30 tablets, a silica gel desiccant, and a polyester fiber coil, and is closed with a child-resistant closure.
Store below 30 °C (86 °F).
Keep container tightly closed.
Dispense only in original container.
USE IN SPECIFIC POPULATIONS SECTION
8 USE IN SPECIFIC POPULATIONS
8.1 Pregnancy
Pregnancy Exposure Registry
There is a pregnancy exposure registry that monitors pregnancy outcomes in individuals exposed to COMPLERA during pregnancy. Healthcare providers are encouraged to register patients on the worldwide Antiretroviral Pregnancy Registry (APR) at www.apregistry.com/.
Risk Summary
Available data from the APR show no increase in the overall risk of major birth defects with first trimester exposure for emtricitabine (FTC), rilpivirine (RPV), or tenofovir (TDF) compared with the background rate for major birth defects of 2.7% in a U.S. reference population of the Metropolitan Atlanta Congenital Defects Program (MACDP) (see Data). In a clinical trial, total rilpivirine exposures were generally lower during pregnancy compared to the postpartum period [see Clinical Pharmacology (12.3)]. The rate of miscarriage for individual drugs is not reported in the APR. The estimated background rate of miscarriage in clinically recognized pregnancies in the U.S. general population is 15–20%.
Based on the experience of HIV-1-infected pregnant individuals who completed a clinical trial through the postpartum period with an RPV-based regimen, no dose adjustments are required for pregnant patients who are already on a stable RPV-containing regimen prior to pregnancy and who are virologically suppressed (HIV-1 RNA less than 50 copies per mL). Lower exposures of RPV were observed during pregnancy, therefore viral load should be monitored closely [see Data and Clinical Pharmacology (12.3)].
In animal studies, no adverse developmental effects were observed when the components of COMPLERA were administered separately during the period of organogenesis at exposures up to 60 and 120 times (mice and rabbits, respectively, FTC) and 15 and 70 times (rats and rabbits, respectively; RPV) the exposure of these components in COMPLERA and at 14 and 19 times (rats and rabbits, respectively) the human dose of TDF based on body surface area comparisons (see Data). Likewise, no adverse developmental effects were seen when FTC was administered to mice and RPV was administered to rats through lactation at exposures up to approximately 60 and 63 times, respectively, the exposure at the recommended daily dose of these components in COMPLERA. No adverse effects were observed in the offspring of rats when TDF was administered through lactation at tenofovir exposures of approximately 14 times the exposure at the recommended daily dosage of COMPLERA.
Data
Human Data
Prospective reports from the APR of overall major birth defects in pregnancies exposed to drug components of COMPLERA are compared with a U.S. background major birth defect rate. Methodological limitations of the APR include the use of MACDP as the external comparator group. Limitations of using an external comparator include differences in methodology and populations, as well as confounding due to the underlying disease.
Emtricitabine: Based on prospective reports to the APR of exposures to FTC- containing regimens during pregnancy resulting in live births (including over 2,750 exposed in the first trimester and over 1,200 exposed in the second/third trimester), there was no increase in overall major birth defects with FTC compared with the background birth defect rate of 2.7% in the U.S. reference population of the MACDP. The prevalence of major birth defects in live births was 2.4% (95% CI: 1.9% to 3.1%) with first trimester exposure to FTC-containing regimens and 2.3% (95% CI: 1.5% to 3.3%) with the second/third trimester exposure to FTC-containing regimens.
Rilpivirine: RPV in combination with a background regimen was evaluated in a clinical trial of 19 HIV-1 infected pregnant subjects on an RPV-based regimen during the second and third trimesters and postpartum. Each of the subjects were on an RPV-based regimen at the time of enrollment. Twelve subjects completed the trial through the postpartum period (6–12 weeks after delivery) and pregnancy outcomes are missing for six subjects. The exposure (C0h and AUC) of total RPV was approximately 30 to 40% lower during pregnancy compared with postpartum (6 to 12 weeks). The protein binding of RPV was similar (>99%) during second trimester, third trimester, and postpartum period [see Clinical Pharmacology (12.3)]. One subject discontinued the trial following fetal death at 25 weeks gestation due to suspected premature rupture of membranes. Among the 12 subjects who were virologically suppressed at baseline (less than 50 copies/mL), virologic response was preserved in 10 subjects (83.3%) through the third trimester visit and in 9 subjects (75%) through the 6–12 week postpartum visit. Virologic outcomes during the third trimester visit were missing for two subjects who were withdrawn (one subject was nonadherent to the study drug and one subject withdrew consent). Among the 10 infants with available HIV test results, all were negative for HIV-1 at the time of delivery and up to 16 weeks postpartum (all 10 infants received prophylactic treatment with zidovudine). RPV was well tolerated during pregnancy and postpartum. There were no new safety findings compared with the known safety profile of RPV in HIV–1-infected adults.
Based on prospective reports to the APR of exposures to RPV-containing regimens during pregnancy (including over 290 exposed during first trimester and over 160 exposed in the second/third trimester), there was no significant increase in overall risk of major birth defects with RPV compared to the background birth defect rate of 2.7% in the U.S. reference population of the MACDP. The prevalence of major birth defects in live births was 1.0% (95% CI: 0.2% to 2.9%) and 1.2% (95% CI: 0.2% to 4.4%) following first and second/third trimester exposure, respectively, to RPV-containing regimens.
Tenofovir DF: Based on prospective reports to the APR of exposures to TDF- containing regimens during pregnancy resulting in live births (including over 3,500 exposed in the first trimester and over 1,500 exposed in the second/third trimester), there was no increase in overall risk of major birth defects compared with the background birth defect rate of 2.7% in the U.S. reference population of the MACDP. The prevalence of major birth defects in live births was 2.3% (95% CI: 1.8% to 2.9%) with first trimester exposure to TDF-containing regimens, and 2.2% (95% CI: 1.6% to 3.1%) with the second/third trimester exposure to TDF-containing regimens.
Animal Data
Emtricitabine: FTC was administered orally to pregnant mice (at 0, 250, 500, or 1,000 mg/kg/day), and rabbits (at 0, 100, 300, or 1,000 mg/kg/day) through organogenesis (on gestation days 6 through 15, and 7 through 19, respectively). No significant toxicological effects were observed in embryo- fetal toxicity studies performed with FTC in mice at exposures (AUC) approximately 60 times higher and in rabbits at approximately 120 times higher than human exposures at the recommended daily dose. In a pre/postnatal development study in mice, FTC was administered orally at doses up to 1,000 mg/kg/day; no significant adverse effects directly related to drug were observed in the offspring exposed daily from before birth (in utero) through sexual maturity at daily exposures (AUC) of approximately 60 times higher than human exposures at the recommended daily dose.
Rilpivirine: RPV was administered orally to pregnant rats (40, 120, or 400 mg/kg/day) and rabbits (5, 10, or 20 mg/kg/day) through organogenesis (on gestation days 6 through 17, and 6 through 19, respectively). No significant toxicological effects were observed in embryo-fetal toxicity studies performed with RPV in rats and rabbits at exposures 15 (rats) and 70 (rabbits) times higher than the exposure in humans at the recommended dose of 25 mg once daily. In a pre/postnatal development study with RPV, where rats were administered up to 400 mg/kg/day through lactation, no significant adverse effects directly related to drug were noted in the offspring.
Tenofovir DF: TDF was administered orally to pregnant rats (at 0, 50, 150, or 450 mg/kg/day) and rabbits (at 0, 30, 100, or 300 mg/kg/day) through organogenesis (on gestation days 7 through 17, and 6 through 18, respectively). No significant toxicological effects were observed in embryo- fetal toxicity studies performed with TDF in rats at doses up to 14 times the human dose based on body surface area comparisons and in rabbits at doses up to 19 times the human dose based on body surface area comparisons. In a pre/postnatal development study in rats, TDF was administered orally through lactation at doses up to 600 mg/kg/day; no adverse effects were observed in the offspring at tenofovir exposures of approximately 2.7 times higher than human exposures at the recommended daily dose of COMPLERA.
8.2 Lactation
Risk Summary
The Centers for Disease Control and Prevention recommend that HIV infected mothers not breastfeed their infants to avoid risking postnatal transmission of HIV.
Based on published data, FTC and tenofovir have been shown to be present in human milk. There are no data on the presence of RPV in human milk. RPV has been shown to be present in rat milk (see Data).
It is not known if the components of COMPLERA affect milk production or have effects on the breastfed child. Because of the potential for: (1) HIV transmission (in HIV-negative infants); (2) developing viral resistance (in HIV-positive infants); and (3) adverse reactions in a breastfed infant similar to those seen in adults, instruct mothers not to breastfeed if they are receiving COMPLERA.
Data
Rilpivirine: In animals, no studies have been conducted to assess the excretion of RPV directly; however RPV was measured in rat pups which were exposed through the milk of treated dams (dosed up to 400 mg/kg/day).
8.4 Pediatric Use
The safety and effectiveness of COMPLERA as a complete regimen for the treatment of HIV-1 infection was established in pediatric subjects 12 years of age and older with body weight greater than or equal to 35 kg [see Dosage and Administration (2.2)]. Use of COMPLERA in this age group weighing at least 35 kg is supported by adequate and well-controlled studies of RPV+FTC+TDF in adults with HIV-1 infection as well as data from pediatric studies of the individual components of COMPLERA (RPV, FTC, and TDF) [see Clinical Pharmacology (12.3), and Clinical Studies (14.2)].
COMPLERA should only be administered to pediatric patients with a body weight greater than or equal to 35 kg. Because COMPLERA is a fixed-dose combination tablet, the dose of COMPLERA cannot be adjusted for patients of lower weight. Safety and effectiveness for COMPLERA have not been established in pediatric patients weighing less than 35 kg [see Adverse Reactions (6.1) and Clinical Pharmacology (12.3)].
8.5 Geriatric Use
Clinical studies of FTC, RPV, or TDF did not include sufficient numbers of subjects aged 65 and over to determine whether they respond differently from younger subjects. In general, dose selection for elderly patients should be cautious, keeping in mind the greater frequency of decreased hepatic, renal, or cardiac function, and of concomitant disease or other drug therapy [see Clinical Pharmacology (12.3)].
8.6 Renal Impairment
Because COMPLERA is a fixed-dose combination, and cannot be dose adjusted, it is not recommended in patients with moderate, severe, or end-stage renal impairment (estimated creatinine clearance below 50 mL per minute) or that require dialysis [see Warnings and Precautions (5.5) and Clinical Pharmacology (12.3)].
8.7 Hepatic Impairment
No dose adjustment of COMPLERA is required in patients with mild (Child-Pugh Class A) or moderate (Child-Pugh Class B) hepatic impairment. COMPLERA has not been studied in patients with severe hepatic impairment (Child-Pugh Class C) [see Clinical Pharmacology (12.3)].
- Pregnancy: Monitor viral load closely during pregnancy as rilpivirine exposures were generally lower during pregnancy. (2.3, 8.1, 12.3)
- Lactation: Breastfeeding not recommended due to the potential for HIV-1 transmission. (8.2)
- Pediatrics: Not recommended for patients weighing less than 35 kg. (8.4)
OVERDOSAGE SECTION
10 OVERDOSAGE
If overdose occurs the patient must be monitored for evidence of toxicity. Treatment of overdose with COMPLERA consists of general supportive measures, including monitoring of vital signs and ECG (QT interval) as well as observation of the clinical status of the patient.
Emtricitabine: Hemodialysis treatment removes approximately 30% of the FTC dose over a 3-hour dialysis period starting within 1.5 hours of FTC dosing (blood flow rate of 400 mL per minute and a dialysate flow rate of 600 mL per minute). It is not known whether FTC can be removed by peritoneal dialysis.
Rilpivirine: There is no specific antidote for overdose with RPV. Human experience of overdose with RPV is limited. Since RPV is highly bound to plasma protein, dialysis is unlikely to result in significant removal of RPV.
Tenofovir DF: Tenofovir is efficiently removed by hemodialysis with an extraction coefficient of approximately 54%. Following a single 300 mg dose of TDF, a 4-hour hemodialysis session removed approximately 10% of the administered tenofovir dose.
DESCRIPTION SECTION
11 DESCRIPTION
COMPLERA is a fixed-dose combination tablet containing FTC, rilpivirine hydrochloride, and TDF. Emtricitabine (FTC) is a synthetic nucleoside analog of cytidine. Rilpivirine (RPV) is a non-nucleoside reverse transcriptase inhibitor. Tenofovir disoproxil fumarate (TDF) is converted in vivo to tenofovir, an acyclic nucleoside phosphonate (nucleotide) analog of adenosine 5′-monophosphate.
COMPLERA tablets are for oral administration. Each tablet contains 200 mg of FTC, 27.5 mg of rilpivirine hydrochloride (equivalent to 25 mg of RPV), and 300 mg of TDF (equivalent to 245 mg of tenofovir disoproxil) as active ingredients. The tablets include the following inactive ingredients: croscarmellose sodium, lactose monohydrate, magnesium stearate, microcrystalline cellulose, polysorbate 20, povidone, pregelatinized starch. The tablets are film coated with a coating material containing hypromellose, lactose monohydrate, polyethylene glycol, titanium dioxide, triacetin.
Emtricitabine: The chemical name of FTC is 5-fluoro-1-[(2R,5S)-2-(hydroxymethyl)-1,3-oxathiolan-5-yl]cytosine. Emtricitabine is the (-) enantiomer of a thio analog of cytidine, which differs from other cytidine analogs in that it has a fluorine in the 5-position.
It has a molecular formula of C8H10FN3O3S and a molecular weight of 247.24. It has the following structural formula:
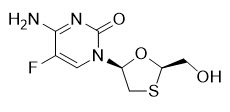
FTC is a white to off-white crystalline powder with a solubility of approximately 112 mg per mL in water at 25 °C.
Rilpivirine: RPV is available as the hydrochloride salt. The chemical name for rilpivirine hydrochloride is 4-[[4-[[4-[(E)-2-cyanoethenyl]-2,6-dimethylphenyl]amino]-2-pyrimidinyl]amino]benzonitrile monohydrochloride. Its molecular formula is C22H18N6 ∙ HCl and its molecular weight is 402.88. Rilpivirine hydrochloride has the following structural formula:
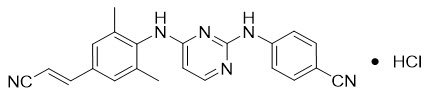
Rilpivirine hydrochloride is a white to almost white powder. Rilpivirine hydrochloride is practically insoluble in water over a wide pH range.
Tenofovir DF: TDF is a fumaric acid salt of the bis- isopropoxycarbonyloxymethyl ester derivative of tenofovir. The chemical name of TDF is 9-[(R)-2 [[bis[[(isopropoxycarbonyl)oxy]- methoxy]phosphinyl]methoxy]propyl]adenine fumarate (1:1). It has a molecular formula of C19H30N5O10P ∙ C4H4O4 and a molecular weight of 635.52. It has the following structural formula:
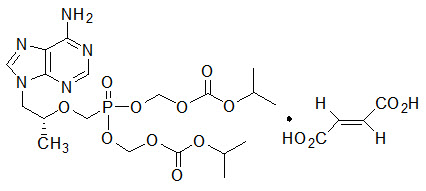
TDF is a white to off-white crystalline powder with a solubility of 13.4 mg per mL in water at 25 °C. All dosages are expressed in terms of TDF except where otherwise noted.
NONCLINICAL TOXICOLOGY SECTION
13 NONCLINICAL TOXICOLOGY
13.1 Carcinogenesis, Mutagenesis, Impairment of Fertility
Emtricitabine: In long-term carcinogenicity studies of FTC, no drug-related increases in tumor incidence were found in mice at doses up to 750 mg per kg per day (26 times the human systemic exposure at the therapeutic dose of 200 mg per day) or in rats at doses up to 600 mg per kg per day (31 times the human systemic exposure at the therapeutic dose).
FTC was not genotoxic in the reverse mutation bacterial test (Ames test), or the mouse lymphoma or mouse micronucleus assays.
FTC did not affect fertility in male rats at approximately 140-fold or in male and female mice at approximately 60-fold higher exposures (AUC) than in humans given the recommended 200 mg daily dose. Fertility was normal in the offspring of mice exposed daily from before birth (in utero) through sexual maturity at daily exposures (AUC) of approximately 60-fold higher than human exposures at the recommended 200 mg daily dose.
Rilpivirine: RPV was evaluated for carcinogenic potential by oral gavage administration to mice and rats up to 104 weeks. Daily doses of 20, 60, and 160 mg per kg per day were administered to mice and doses of 40, 200, 500, and 1500 mg per kg per day were administered to rats. In rats, there were no drug- related neoplasms. In mice, RPV was positive for hepatocellular neoplasms in both males and females. The observed hepatocellular findings in mice may be rodent-specific. At the lowest tested doses in the carcinogenicity studies, the systemic exposures (based on AUC) to RPV were 21 fold (mice) and 3 fold (rats), relative to those observed in humans at the recommended dose (25 mg once daily).
RPV has tested negative in the absence and presence of a metabolic activation system, in the in vitro Ames reverse mutation assay and in vitro clastogenicity mouse lymphoma assay. RPV did not induce chromosomal damage in the in vivo micronucleus test in mice.
In a study conducted in rats, there were no effects on mating or fertility with RPV up to 400 mg per kg per day, a dose of RPV that showed maternal toxicity. This dose is associated with an exposure that is approximately 40 times higher than the exposure in humans at the recommended dose of 25 mg once daily.
Tenofovir DF: Long-term oral carcinogenicity studies of TDF in mice and rats were carried out at exposures up to approximately 16 times (mice) and 5 times (rats) those observed in humans at the therapeutic dose for HIV-1 infection. At the high dose in female mice, liver adenomas were increased at exposures 16 times that in humans. In rats, the study was negative for carcinogenic findings at exposures up to 5 times that observed in humans at the therapeutic dose.
Tenofovir DF was mutagenic in the in vitro mouse lymphoma assay and negative in an in vitro bacterial mutagenicity test (Ames test). In an in vivo mouse micronucleus assay, TDF was negative when administered to male mice.
There were no effects on fertility, mating performance, or early embryonic development when TDF was administered to male rats at a dose equivalent to 10 times the human dose based on body surface area comparisons for 28 days prior to mating and to female rats for 15 days prior to mating through day 7 of gestation. There was, however, an alteration of the estrous cycle in female rats.
13.2 Animal Toxicology and/or Pharmacology
Tenofovir DF: Tenofovir and TDF administered in toxicology studies to rats, dogs, and monkeys at exposures (based on AUCs) greater than or equal to 6-fold those observed in humans caused bone toxicity. In monkeys the bone toxicity was diagnosed as osteomalacia. Osteomalacia observed in monkeys appeared to be reversible upon dose reduction or discontinuation of tenofovir. In rats and dogs, the bone toxicity manifested as reduced bone mineral density. The mechanism(s) underlying bone toxicity is unknown.
Evidence of renal toxicity was noted in 4 animal species. Increases in serum creatinine, BUN, glycosuria, proteinuria, phosphaturia, and/or calciuria and decreases in serum phosphate were observed to varying degrees in these animals. These toxicities were noted at exposures (based on AUCs) 2–20 times higher than those observed in humans. The relationship of the renal abnormalities, particularly the phosphaturia, to the bone toxicity is not known.
CLINICAL STUDIES SECTION
14 CLINICAL STUDIES
14.1 Adult Subjects
In HIV-1-Infected Adult Subjects With No Antiretroviral Treatment History
The efficacy of COMPLERA is based on the analyses of 48- and 96-week data from two randomized, double-blind, controlled studies (Study C209 [ECHO] and TRUVADA subset of Study C215 [THRIVE]) in treatment-naïve, HIV-1-infected subjects (N=1368). The studies are identical in design with the exception of the background regimen (BR). Subjects were randomized in a 1:1 ratio to receive either RPV 25 mg (N=686) once daily or EFV 600 mg (N=682) once daily in addition to a BR. In Study C209 (N=690), the BR was FTC/TDF. In Study C215 (N=678), the BR consisted of 2 NRTIs: FTC/TDF (60%, n=406), lamivudine/zidovudine (30%, n=204), or abacavir + lamivudine (10%, n=68).
For subjects who received FTC/TDF (N=1096) in studies C209 and C215, the mean age was 37 years (range 18–78), 78% were male, 62% were White, 24% were Black, and 11% were Asian. The mean baseline CD4+ cell count was 265 cells/mm3 (range 1–888) and 31% had CD4+ cell counts <200 cells/mm3. The median baseline plasma HIV-1 RNA was 5 log10 copies/mL (range 2–7). Subjects were stratified by baseline HIV-1 RNA. Fifty percent of subjects had baseline viral load ≤100,000 copies/mL, 39% of subjects had baseline viral load between 100,000 copies/mL to 500,000 copies/mL, and 11% of subjects had baseline viral load >500,000 copies/mL.
Treatment outcomes through 96 weeks for the subset of subjects receiving FTC/TDF in studies C209 and C215 (Table 16) are generally consistent with treatment outcomes for all participating subjects (presented in the prescribing information for Edurant). The incidence of virologic failure was higher in the RPV arm than the EFV arm at Week 96. Virologic failures and discontinuations due to adverse events mostly occurred in the first 48 weeks of treatment.
Table 16 Pooled Virologic Outcome of Randomized Treatment of Studies C209 and C215 at Week 96 in Adult Subjects With No Antiviral Treatment History in Combination with FTC/TDF) at Week 96*|
RPV + FTC/TDF |
EFV + FTC/TDF | |
|---|---|---|
|
N=550 |
N=546 | |
| ||
|
HIV-1 RNA <50 copies/mL**†** |
77% |
77% |
|
HIV-1 RNA ≥50 copies/mL**‡** |
14% |
8% |
|
No Virologic Data at Week 96 Window | ||
|
Discontinued study due to adverse event or death§ |
4% |
9% |
|
Discontinued study for other reasons¶ and the last available HIV-1 RNA <50 copies/mL (or missing) |
4% |
6% |
|
Missing data during window but on study |
<1% |
<1% |
|
HIV-1 RNA <50 copies/mL | ||
|
≤100,000 |
83% |
80% |
|
71% |
74% |
|
HIV-1 RNA ≥50 copies/mL**‡****** | ||
|
≤100,000 |
7% |
5% |
|
22% |
12% |
|
HIV-1 RNA <50 copies/mL | ||
|
<200 |
68% |
72% |
|
≥200 |
82% |
79% |
|
HIV-1 RNA ≥50 copies/mL**‡****** | ||
|
<200 |
27% |
12% |
|
≥200 |
8% |
7% |
Based on the pooled data from studies C209 and C215, the mean CD4+ cell count increase from baseline at Week 96 was 226 cells/mm3 for RPV + FTC/TDF-treated subjects and 223 cells/mm3 for EFV + FTC/TDF-treated subjects.
In Virologically Suppressed HIV-1-Infected Adult Subjects
The efficacy and safety of switching from a ritonavir-boosted protease inhibitor in combination with two NRTIs to COMPLERA was evaluated in Study 106, a randomized, open-label study in virologically suppressed HIV-1-infected adults. Subjects had to be on either their first or second antiretroviral regimen with no history of virologic failure, have no current or past history of resistance to any of the three components of COMPLERA, and must have been suppressed (HIV-1 RNA <50 copies/mL) for at least 6 months prior to screening. Subjects were randomized in a 2:1 ratio to either switch to COMPLERA at baseline (COMPLERA arm, N=317), or stay on their baseline antiretroviral regimen for 24 weeks (SBR arm, N=159) and then switch to COMPLERA for an additional 24 weeks (N=52). Subjects had a mean age of 42 years (range 19–73), 88% were male, 77% were White, 17% were Black, and 17% were Hispanic/Latino. The mean baseline CD4+ cell count was 584 cells/mm3 (range 42–1484). Randomization was stratified by use of TDF and/or lopinavir/ritonavir in the baseline regimen.
Treatment outcomes are presented in Table 17.
Table 17 Virologic Outcomes of Study GS-US-264-0106 in Virologically Suppressed Subjects|
COMPLERA |
Stayed on Baseline Regimen (SBR) | |
|---|---|---|
|
Week 48* |
Week 24† | |
|
N=317 |
N=159 | |
| ||
|
HIV-1 RNA <50 copies/mL‡ |
89% (283/317) |
90% (143/159) |
|
HIV-1 RNA ≥50 copies/mL§ |
3% (8/317) |
5% (8/159) |
|
No Virologic Data at Week 24 Window | ||
|
Discontinued study drug due to AE or death¶ |
2% (7/317) |
0% |
|
Discontinued study drug due to other reasons and last available HIV-1 RNA <50 copies/mL# |
5% (16/317) |
3% (5/159) |
|
Missing data during window but on study drug |
1% (3/317) |
2% (3/159) |
14.2 Pediatric Subjects
The pharmacokinetics, safety, and efficacy of RPV in combination with other antiretroviral agents was evaluated in a single-arm, open-label Phase 2 trial in antiretroviral treatment-naïve HIV-1-infected pediatric subjects 12 to less than 18 years of age and weighing at least 32 kg (TMC-C213). Thirty-six (36) subjects were enrolled with a median age of 14.5 years (range 12 to 17 years), and were 55.6% female, 88.9% Black, and 11.1% Asian. The majority of subjects (24/36) received RPV in combination with FTC and TDF. Of these 24 subjects, 20 had baseline HIV RNA ≤100,000 copies/mL. The baseline characteristics and efficacy outcomes at Week 48 are further described below for the 20 subjects.
The median baseline plasma HIV-1 RNA and CD4+ cell count were 49,550 (range 2060 to 92,600 copies/mL) and 437.5 cells/mm3 (range 123 to 983 cells/mm3), respectively. At Week 48, 80% (16/20) of the subjects had HIV RNA <50 copies/mL, 15% (3/20) had HIV RNA ≥50 copies/mL, and one subject discontinued therapy prior to Week 48 and before reaching virologic suppression (HIV RNA <50 copies/mL). At Week 48, the mean increase in CD4+ cell count from baseline was 225 cells/mm3.
INFORMATION FOR PATIENTS SECTION
17 PATIENT COUNSELING INFORMATION
Advise the patient to read the approved patient labeling (Patient Information).
Posttreatment Acute Exacerbation of Hepatitis B in Patients Coinfected with HIV-1 and HBV
Severe acute exacerbations of hepatitis B have been reported in patients who are coinfected with HBV and HIV-1 and who have discontinued products containing FTC or TDF [see Warnings and Precautions (5.1)]. Advise patients to not discontinue COMPLERA without first informing their healthcare provider.
Severe Skin Reactions and Hypersensitivity
Advise patients to immediately contact their healthcare provider if they develop a rash. Instruct patients to immediately stop taking COMPLERA and seek medical attention if they develop a rash associated with any of the following symptoms, as it may be a sign of a more serious reaction such as DRESS severe hypersensitivity: fever, blisters, mucosal involvement, eye inflammation (conjunctivitis), severe allergic reaction causing swelling of the face, eyes, lips, mouth, tongue, or throat which may lead to difficulty swallowing or breathing, and any signs and symptoms of liver problems, as they may be a sign of a more serious reaction. Patients should understand that if severe rash occurs, they will be closely monitored, laboratory tests will be performed and appropriate therapy will be initiated [see Warnings and Precautions (5.2)].
Hepatotoxicity
Inform patients that hepatotoxicity has been reported with COMPLERA and that monitoring for hepatotoxicity is recommended [see Warnings and Precautions (5.3)].
Depressive Disorders
Inform patients that depressive disorders (depressed mood, depression, dysphoria, major depression, mood altered, negative thoughts, suicide attempt, suicidal ideation) have been reported with COMPLERA. Advise patients to seek immediate medical evaluation if they experience depressive symptoms [see Warnings and Precautions (5.4)].
New Onset or Worsening Renal Impairment
Inform patients that renal impairment, including cases of acute renal failure and Fanconi syndrome, has been reported in association with the use of TDF. COMPLERA should be avoided with concurrent or recent use of a nephrotoxic agent (e.g., high-dose or multiple NSAIDs) [see Warnings and Precautions (5.5)].
Bone Loss and Mineralization Defects
Inform patients that decreases in bone mineral density have been observed with the use of TDF. Assessment of bone mineral density (BMD) should be considered in patients who have a history of pathologic bone fracture or other risk factors for osteoporosis or bone loss [see Warnings and Precautions (5.6)].
Drug Interactions
COMPLERA may interact with many drugs and is not recommended to be coadministered with numerous drugs. Advise patients to report to their healthcare provider the use of any other prescription or nonprescription medication or herbal products, including St. John's wort [see Contraindications (4), Warnings and Precautions (5.7), and Drug Interactions (7)].
For patients receiving rifabutin, an additional 25 mg tablet of RPV (Edurant) once per day is recommended to be taken concomitantly with COMPLERA and with a meal for the duration of rifabutin coadministration.
Lactic Acidosis and Severe Hepatomegaly
Inform patients that lactic acidosis and severe hepatomegaly with steatosis, including fatal cases, have been reported. Treatment with COMPLERA should be suspended in any patient who develops clinical symptoms suggestive of lactic acidosis or pronounced hepatotoxicity [see Warnings and Precautions (5.8)].
Immune Reconstitution Syndrome
Inform patients to inform their healthcare provider immediately of any signs and symptoms of inflammation from previous infections, which may occur soon after anti-HIV treatment is started [see Warnings and Precautions (5.9)].
Dosing Instructions
Advise patients that it is important to take COMPLERA on a regular dosing schedule with food and to avoid missing doses. A protein drink is not a substitute for food. If the healthcare provider decides to stop COMPLERA and the patient is switched to new medicines to treat HIV that include RPV tablets, the RPV tablets should be taken only with a meal.
Pregnancy Registry
Inform patients that there is an antiretroviral pregnancy registry to monitor fetal outcomes in those exposed to COMPLERA during pregnancy [see Use in Specific Populations (8.1)].
Lactation
Instruct patients with HIV-1 infection not to breastfeed because HIV-1 can be passed to the baby in breast milk [see Use in Specific Populations (8.2)].