
REPATHA
These highlights do not include all the information needed to use REPATHA safely and effectively. See full prescribing information for REPATHA. REPATHA (evolocumab) injection, for subcutaneous use Initial U.S. Approval: 2015
cd61e902-166d-4aa6-9f3c-a18c1008d07e
HUMAN PRESCRIPTION DRUG LABEL
Jan 25, 2024
Amgen USA Inc.
DUNS: 962075045
Products 5
Detailed information about drug products covered under this FDA approval, including NDC codes, dosage forms, ingredients, and administration routes.
Evolocumab
Product Details
FDA regulatory identification and product classification information
FDA Identifiers
Product Classification
Product Specifications
INGREDIENTS (6)
Evolocumab
Product Details
FDA regulatory identification and product classification information
FDA Identifiers
Product Classification
Product Specifications
INGREDIENTS (6)
Evolocumab
Product Details
FDA regulatory identification and product classification information
FDA Identifiers
Product Classification
Product Specifications
Evolocumab
Product Details
FDA regulatory identification and product classification information
FDA Identifiers
Product Classification
Product Specifications
INGREDIENTS (6)
Evolocumab
Product Details
FDA regulatory identification and product classification information
FDA Identifiers
Product Classification
Product Specifications
INGREDIENTS (6)
Drug Labeling Information
PACKAGE LABEL.PRINCIPAL DISPLAY PANEL
PRINCIPAL DISPLAY PANEL - 140 mg/mL Syringe Carton - NDC 72511-501-01
1 x 1 mL Prefilled Syringe
NDC 72511-501-01
AMGEN®
140
mg/mL
Repatha®
(evolocumab)
Injection
Not made
with natural
rubber latex
140 mg/mL
Prefilled Syringe
For Subcutaneous Use Only
Store refrigerated at 2°C to 8°C (36°F to 46°F). Do Not Freeze or Shake.
Store in Carton to Protect from Light.
(see side panel for additional storage information)
Sterile Solution – No Preservative
Rx Only
Do not re-use
CAUTION, See package insert for full prescribing
information and Instructions for Use
Keep out of the sight and reach of children
GTIN
SN
LOT
Exp.

INDICATIONS & USAGE SECTION
1 INDICATIONS AND USAGE
REPATHA is indicated:
- To reduce the risk of major adverse cardiovascular (CV) events (CV death, myocardial infarction, stroke, unstable angina requiring hospitalization, or coronary revascularization) in adults with established cardiovascular disease
- As an adjunct to diet, alone or in combination with other low-density lipoprotein cholesterol (LDL-C)-lowering therapies, in adults with primary hyperlipidemia, including heterozygous familial hypercholesterolemia (HeFH), to reduce LDL-C
- As an adjunct to diet and other LDL-C-lowering therapies in pediatric patients aged 10 years and older with HeFH, to reduce LDL-C
- As an adjunct to other LDL-C-lowering therapies in adults and pediatric patients aged 10 years and older with homozygous familial hypercholesterolemia (HoFH), to reduce LDL-C
REPATHA is a PCSK9 (proprotein convertase subtilisin kexin type 9) inhibitor indicated:
- To reduce the risk of major adverse cardiovascular (CV) events (CV death, myocardial infarction, stroke, unstable angina requiring hospitalization, or coronary revascularization) in adults with established cardiovascular disease (1)
- as an adjunct to diet, alone or in combination with other low-density lipoprotein cholesterol (LDL-C)-lowering therapies, in adults with primary hyperlipidemia, including heterozygous familial hypercholesterolemia (HeFH), to reduce LDL-C (1)
- as an adjunct to diet and other LDL-C-lowering therapies in pediatric patients aged 10 years and older with HeFH, to reduce LDL-C (1)
- as an adjunct to other LDL-C-lowering therapies in adults and pediatric patients aged 10 years and older with homozygous familial hypercholesterolemia (HoFH), to reduce LDL-C (1)
CONTRAINDICATIONS SECTION
4 CONTRAINDICATIONS
REPATHA is contraindicated in patients with a history of a serious hypersensitivity reaction to evolocumab or any of the excipients in REPATHA. Serious hypersensitivity reactions including angioedema have occurred in patients treated with REPATHA [see Warnings and Precautions (5.1)].
Patients with a history of a serious hypersensitivity reaction to evolocumab or any of the excipients in REPATHA. (4)
WARNINGS AND PRECAUTIONS SECTION
5 WARNINGS AND PRECAUTIONS
5.1 Hypersensitivity Reactions
Hypersensitivity reactions, including angioedema, have been reported in patients treated with REPATHA. If signs or symptoms of serious hypersensitivity reactions occur, discontinue treatment with REPATHA, treat according to the standard of care, and monitor until signs and symptoms resolve. REPATHA is contraindicated in patients with a history of serious hypersensitivity reactions to evolocumab or any excipient in REPATHA [see Contraindications (4)].
The prefilled single-dose SureClick® autoinjector and prefilled single-dose syringe presentations of REPATHA that contain dry natural rubber (a derivative of latex) in the needle cover may cause an allergic reaction in individuals sensitive to latex. Instruct patients to inform their healthcare provider if they are sensitive to latex. Consider prescribing a presentation of REPATHA that does not contain dry natural rubber for individuals that are sensitive to latex [see How Supplied/Storage and Handling (16)].
Hypersensitivity Reactions: Angioedema has occurred. If signs or symptoms of serious hypersensitivity reactions occur, discontinue treatment with REPATHA, treat according to the standard of care, and monitor until signs and symptoms resolve. (5.1)
ADVERSE REACTIONS SECTION
6 ADVERSE REACTIONS
The following adverse reactions are also discussed in other sections of the label:
- Hypersensitivity Reactions [see Warnings and Precautions (5.1)]
6.1 Clinical Trials Experience
Because clinical trials are conducted under widely varying conditions, adverse reaction rates observed in the clinical trials of a drug cannot be directly compared to rates in the clinical trials of another drug and may not reflect the rates observed in clinical practice.
Adverse Reactions in Adults with Primary Hyperlipidemia
The data described below reflect exposure to REPATHA in 8 placebo-controlled trials that included 2651 patients treated with REPATHA, including 557 exposed for 6 months and 515 exposed for 1 year (median treatment duration of 12 weeks). The mean age of the population was 57 years, 49% of the population were women, 85% White, 6% Black, 8% Asians, and 2% other races.
Adverse Reactions in a 52-Week Controlled Trial
In a 52-week, double-blind, randomized, placebo-controlled trial, 599 patients received 420 mg of REPATHA subcutaneously once monthly [see Clinical Studies (14)]. The mean age was 56 years (range: 22 to 75 years), 23% were older than 65 years, 52% women, 80% White, 8% Black, 6% Asian; 6% identified as Hispanic ethnicity. Adverse reactions reported in at least 3% of REPATHA-treated patients, and more frequently than in placebo-treated patients are shown in Table 1. Adverse reactions led to discontinuation of treatment in 2.2% of REPATHA-treated patients and 1% of placebo-treated patients. The most common adverse reaction that led to REPATHA treatment discontinuation and occurred at a rate greater than placebo was myalgia (0.3% versus 0% for REPATHA and placebo, respectively).
Table 1. Adverse Reactions Occurring in ≥ 3% of REPATHA-treated Patients and More Frequently than with Placebo in a 52-Week Trial|
Placebo |
REPATHA | |
|---|---|---|
| ||
|
Nasopharyngitis |
9.6 |
10.5 |
|
Upper respiratory tract infection |
6.3 |
9.3 |
|
Influenza |
6.3 |
7.5 |
|
Back pain |
5.6 |
6.2 |
|
Injection site reactions* |
5.0 |
5.7 |
|
Cough |
3.6 |
4.5 |
|
Urinary tract infection |
3.6 |
4.5 |
|
Sinusitis |
3.0 |
4.2 |
|
Headache |
3.6 |
4.0 |
|
Myalgia |
3.0 |
4.0 |
|
Dizziness |
2.6 |
3.7 |
|
Musculoskeletal pain |
3.0 |
3.3 |
|
Hypertension |
2.3 |
3.2 |
|
Diarrhea |
2.6 |
3.0 |
|
Gastroenteritis |
2.0 |
3.0 |
Adverse Reactions in Seven Pooled 12-Week Controlled Trials
In seven pooled 12-week, double-blind, randomized, placebo-controlled trials, 993 patients received 140 mg of REPATHA subcutaneously every 2 weeks and 1059 patients received 420 mg of REPATHA subcutaneously monthly. The mean age was 57 years (range: 18 to 80 years), 29% were older than 65 years, 49% women, 85% White, 5% Black, 9% Asian; 5% identified as Hispanic ethnicity. Adverse reactions reported in at least 1% of REPATHA-treated patients, and more frequently than in placebo-treated patients, are shown in Table 2.
Table 2. Adverse Reactions Occurring in ≥ 1% of REPATHA-treated Patients and More Frequently than with Placebo in Pooled 12-Week Trials|
Placebo |
REPATHA* | |
|---|---|---|
| ||
|
Nasopharyngitis |
3.9 |
4.0 |
|
Back pain |
2.2 |
2.3 |
|
Upper respiratory tract infection |
2.0 |
2.1 |
|
Arthralgia |
1.6 |
1.8 |
|
Nausea |
1.2 |
1.8 |
|
Fatigue |
1.0 |
1.6 |
|
Muscle spasms |
1.2 |
1.3 |
|
Urinary tract infection |
1.2 |
1.3 |
|
Cough |
0.7 |
1.2 |
|
Influenza |
1.1 |
1.2 |
|
Contusion |
0.5 |
1.0 |
Adverse Reactions in Eight Pooled Controlled Trials (Seven 12-Week Trials and One 52-Week Trial)
The adverse reactions described below are from a pool of the 52-week trial and seven 12-week trials. The mean and median exposure durations of REPATHA in this pool of eight trials were 20 weeks and 12 weeks, respectively.
Local Injection Site Reactions
Injection site reactions occurred in 3.2% and 3.0% of REPATHA-treated and placebo-treated patients, respectively. The most common injection site reactions were erythema, pain, and bruising. The proportions of patients who discontinued treatment due to local injection site reactions in REPATHA- treated patients and placebo-treated patients were 0.1% and 0%, respectively.
Hypersensitivity Reactions
Hypersensitivity reactions occurred in 5.1% and 4.7% of REPATHA-treated and placebo-treated patients, respectively. The most common hypersensitivity reactions were rash (1.0% versus 0.5% for REPATHA and placebo, respectively), eczema (0.4% versus 0.2%), erythema (0.4% versus 0.2%), and urticaria (0.4% versus 0.1%).
Adverse Reactions in the Cardiovascular Outcomes Trial
In a double-blind, randomized, placebo-controlled cardiovascular outcomes trial, 27,525 patients received at least one dose of REPATHA or placebo [see Clinical Studies (14)]. The mean age was 62.5 years (range: 40 to 86 years), 45% were 65 years or older, 9% were 75 years or older, 25% women, 85% White, 2% Black and 10% Asian; 8% identified as Hispanic ethnicity. Patients were exposed to REPATHA or placebo for a median of 24.8 months; 91% of patients were exposed for ≥ 12 months, 54% were exposed for ≥ 24 months and 5% were exposed for ≥ 36 months.
The safety profile of REPATHA in this trial was generally consistent with the safety profile described above in the 12- and 52-week controlled trials involving patients with primary hyperlipidemia. Common adverse reactions (> 5% of patients treated with REPATHA and occurring more frequently than placebo) included diabetes mellitus (8.8% REPATHA, 8.2% placebo), nasopharyngitis (7.8% REPATHA, 7.4% placebo), and upper respiratory tract infection (5.1% REPATHA, 4.8% placebo).
Among the 16,676 patients without diabetes mellitus at baseline, the incidence of new-onset diabetes mellitus during the trial was 8.1% in patients treated with REPATHA compared with 7.7% in patients that received placebo.
Adverse Reactions in Pediatric Patients with HeFH
In a 24-week, randomized, placebo-controlled, double-blind trial of 157 pediatric patients with HeFH, 104 patients received 420 mg REPATHA subcutaneously once monthly [see Clinical Studies (14)]. The mean age was 13.7 years (range: 10 to 17 years), 56% were female, 85% White, 1% Black, 1% Asian, and 13% other; 8% identified as Hispanic ethnicity. Common adverse reactions (> 5% of patients treated with REPATHA and occurring more frequently than placebo) included:
- Nasopharyngitis (12% versus 11%)
- Headache (11% versus 2%)
- Oropharyngeal pain (7% versus 0%)
- Influenza (6% versus 4%)
- Upper respiratory tract infection (6% versus 2%)
Adverse Reactions in Adults and Pediatric Patients with HoFH
In a 12-week, double-blind, randomized, placebo-controlled trial of 49 patients with HoFH, 33 patients received 420 mg of REPATHA subcutaneously once monthly [see Clinical Studies (14)]. The mean age was 31 years (range: 13 to 57 years), 49% were women, 90% White, 4% Asian, and 6% other. The adverse reactions that occurred in at least two (6.1%) REPATHA-treated patients, and more frequently than in placebo-treated patients, included:
- Upper respiratory tract infection (9.1% versus 6.3%)
- Influenza (9.1% versus 0%)
- Gastroenteritis (6.1% versus 0%)
- Nasopharyngitis (6.1% versus 0%)
In a multicenter, open-label 5-year extension study, 106 patients with HoFH, including 14 pediatric patients, received 420 mg of REPATHA subcutaneously once monthly or every 2 weeks [see Clinical Studies (14)]. The mean age was 34 years (range: 13 to 68 years), 51% were women, 80% White, 12% Asian, 1% Native American, and 7% other; 5% identified as Hispanic ethnicity. No new adverse reactions were observed during the open-label extension study.
6.2 Immunogenicity
As with all therapeutic proteins, there is potential for immunogenicity. The detection of antibody formation is highly dependent on the sensitivity and specificity of the assay. Additionally, the observed incidence of antibody (including neutralizing antibody) positivity in an assay may be influenced by several factors including assay methodology, sample handling, timing of sample collection, concomitant medications, and underlying disease. For these reasons, comparison of the incidence of antibodies to REPATHA in the studies described below with the incidence of antibodies in other studies or to other products may be misleading.
The immunogenicity of REPATHA has been evaluated using an electrochemiluminescent bridging screening immunoassay for the detection of binding anti-drug antibodies. For patients whose sera tested positive in the screening immunoassay, an in vitro biological assay was performed to detect neutralizing antibodies.
In a pool of placebo- and active-controlled clinical trials, 0.3% (48 out of 17,992) of adult patients treated with at least one dose of REPATHA tested positive for the development of binding antibodies. Patients whose sera tested positive for binding antibodies were further evaluated for neutralizing antibodies; none of the patients tested positive for neutralizing antibodies.
The development of anti-evolocumab antibodies was not detected in clinical trials of pediatric patients treated with REPATHA.
There was no evidence that the presence of anti-drug binding antibodies impacted the pharmacokinetic profile, clinical response, or safety of REPATHA.
6.3 Postmarketing Experience
The following additional adverse reactions have been identified during post- approval use of REPATHA. Because these reactions are reported voluntarily from a population of uncertain size, it is not always possible to reliably estimate their frequency or establish a causal relationship to drug exposure.
- Hypersensitivity reactions: Angioedema
- Influenza-like illness
Common (> 5% of patients treated with REPATHA and more frequently than placebo) adverse reactions in adults with:
Primary hyperlipidemia: nasopharyngitis, upper respiratory tract infection, influenza, back pain, and injection site reactions. (6)
Established CVD: diabetes mellitus, nasopharyngitis and upper respiratory tract infection. (6)
To report SUSPECTED ADVERSE REACTIONS, contact Amgen Medical Information at 1-800-77-AMGEN (1-800-772-6436) or FDA at 1-800-FDA-1088 or www.fda.gov/medwatch.
INSTRUCTIONS FOR USE SECTION
|
Instructions for Use | |
|
Getting to know the prefilled syringe | |
|
Before use |
After use |
|
|
|
|
Needle is inside |
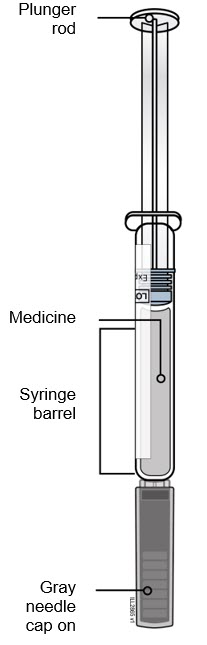

|
Important |
|
Before you use a prefilled single-dose syringe, read this important information:
Storage of REPATHA**:**
|
|
Do not: |
|
*Do not use the prefilled syringe if the packaging is open or damaged. *Do not remove the gray needle cap from the prefilled syringe until you are ready to inject. *Do not use the prefilled syringe if it has been dropped onto a hard surface. Part of the prefilled syringe may be broken even if you cannot see the break. Use a new prefilled syringe and call 1-844-REPATHA (1-844-737-2842). *Do not use the prefilled syringe after the expiration date. |
|
A healthcare provider who knows how to use the prefilled syringe should be
able to answer your questions. For more information, call 1-844-REPATHA
(1-844-737-2842) or visit www.REPATHA.com. |
|
Step 1: Prepare | |
|
1 A |
Remove the prefilled syringe carton from the refrigerator and wait 30 minutes. |
|
Wait at least30 minutes for the prefilled syringe in the carton to reach room temperature before injecting. |
|
|
Using the prefilled syringe at room temperature makes sure the full dose is delivered and allows for a more comfortable injection.Do not heat the syringe. Let it warm up on its own naturally. | |
|
Do not try to warm the prefilled syringe by using a heat source such as hot water or microwave. | |
|
Do not leave the prefilled syringe in direct sunlight. | |
|
Do notshake the prefilled syringe. | |
|
1 B |
Gather all materials needed for your injection. |
|
Wash your hands thoroughly with soap and water. | |
|
On a clean, well-lit, flat work surface, place: | |
| |
|
| |
|
1 C |
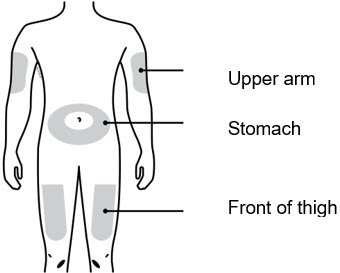
Choose your injection site. |
|
| |
|
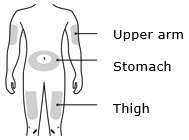
You can use the:
If someone else is giving you the injection, they can also use the outer
area of the upper arm. | |
|
1 D |
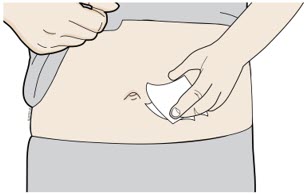
Clean your injection site. |
|
| |
|
Clean your injection site with an alcohol wipe. Let your skin dry before
injecting. | |
|
1 E |
Remove prefilled syringe from tray. |
|
| |
|
To remove:
| |
|
1 F |
Check the medicine and syringe. |
|
| |
|
Always hold the prefilled syringe by the syringe barrel. | |
In any above cases, use a new prefilled syringe and call 1-844-REPATHA (1-844-737-2842) or visit www.REPATHA.com. |

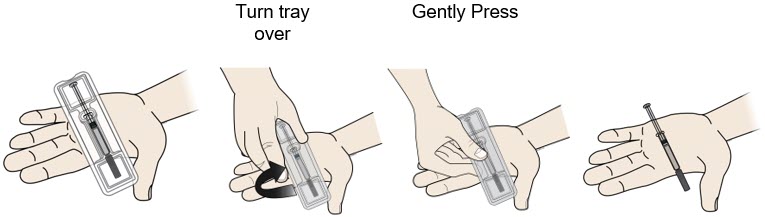

|
Step 2: Get ready | |
|
2 A |
Carefully pull the gray needle cap straight out and away from your body.Do not leave the gray needle cap off for more than5 minutes. This can dry out the medicine. |
|
|
|
|
It is normal to see a drop of medicine at the end of the needle. |
Place the gray needle cap in the sharps disposal container right away. |
|
Do nottwist or bend the gray needle cap. This can damage the needle. | |
|
Do notput the gray needle cap back onto the prefilled syringe. | |
|
Do nottry to remove any air bubbles in the syringe before the injection. |
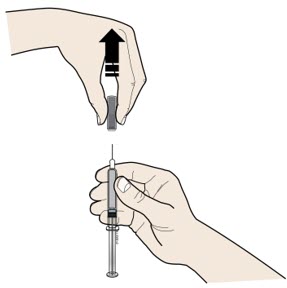
|
2 B |
Pinch your injection site to create a firm surface. |
|
| |
|
Pinch the skin firmly between your thumb and fingers, creating an area about 2 inches wide. | |
|
It is important to keep the skin pinched while injecting. |
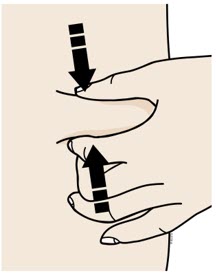
|
Step 3: Inject | |
|
3 A |
Hold thepinch. Insert the needle into the skin using a 45 to 90 degree angle. |
|
| |
|
Do notplace your finger on the plunger rod while inserting the needle. |
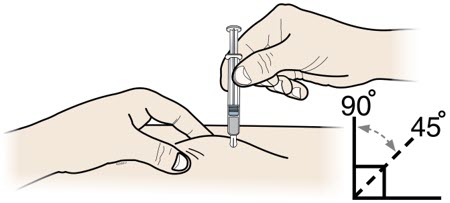
|
3 B |
Using slow and constant pressure,push the plunger rod all the way down until the prefilled syringe is empty. You may have to push harder on the plunger rod than for other injectable medicines. |
|
|
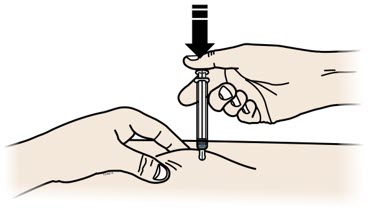
|
3 C |
When the prefilled syringe is empty,release your thumb, and gently lift the syringe out of the skin. |
|
| |
|
Do notput the gray needle cap back onto the used prefilled syringe. |
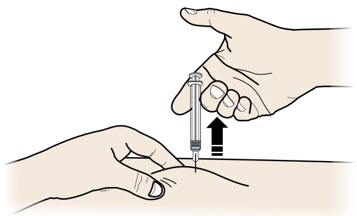
|
Step 4: Finish | |
|
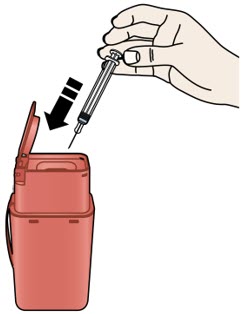
4 A |
Place the used prefilled syringe in a sharps disposal container right away. |
|
| |
|
Do notreuse the used prefilled syringe. | |
|
Do notuse any medicine that is left in the used prefilled syringe.
Do not dispose of your used sharps disposal container in your household trash unless your community guidelines permit this. Do not recycle your used sharps disposal container. | |
|
Keep the used syringe and sharps container out of the sight and reach of children. | |
|
4 B |
Check the injection site. |
|
If there is blood, press a cotton ball or gauze pad on your injection site. Apply an adhesive bandage if needed. | |
|
Do notrub the injection site. |
This Instructions for Use has been approved by the U.S. Food and Drug Administration.
Manufactured by:
Amgen Inc.
Thousand Oaks, CA 91320-1799
U.S. License Number 1080
© 2024 Amgen Inc.
All rights reserved.
USE IN SPECIFIC POPULATIONS SECTION
8 USE IN SPECIFIC POPULATIONS
8.1 Pregnancy
Risk Summary
Available data from clinical trials and postmarketing reports on REPATHA use in pregnant women are insufficient to evaluate for a drug-associated risk of major birth defects, miscarriage or other adverse maternal or fetal outcomes. In animal reproduction studies, there were no effects on pregnancy or neonatal/infant development when monkeys were subcutaneously administered evolocumab from organogenesis through parturition at dose exposures up to 12 times the exposure at the maximum recommended human dose of 420 mg every month. In a similar study with another drug in the PCSK9 inhibitor antibody class, humoral immune suppression was observed in infant monkeys exposed to that drug in utero at all doses. The exposures where immune suppression occurred in infant monkeys were greater than those expected clinically. No assessment for immune suppression was conducted with evolocumab in infant monkeys. Measurable evolocumab serum concentrations were observed in the infant monkeys at birth at comparable levels to maternal serum, indicating that evolocumab, like other IgG antibodies, crosses the placental barrier. Monoclonal antibodies are transported across the placenta in increasing amounts especially near term; therefore, evolocumab has the potential to be transmitted from the mother to the developing fetus.
The estimated background risk of major birth defects and miscarriage for the indicated population(s) is unknown. In the U.S. general population, the estimated background risk of major birth defects and miscarriage in clinically recognized pregnancies is 2-4% and 15-20%, respectively.
There is a pregnancy safety study for REPATHA. If REPATHA is administered during pregnancy, healthcare providers should report REPATHA exposure by contacting Amgen at 1-800-77-AMGEN (1-800-772-6436) or https://wwwext.amgen.com/products/global-patient-safety/adverse-event- reporting.
Data
Animal Data
In cynomolgus monkeys, no effects on embryo-fetal or postnatal development (up to 6 months of age) were observed when evolocumab was dosed during organogenesis to parturition at 50 mg/kg once every 2 weeks by the subcutaneous route at exposures 30- and 12-fold the recommended human doses of 140 mg every 2 weeks and 420 mg once monthly, respectively, based on plasma AUC. No test of humoral immunity in infant monkeys was conducted with evolocumab.
8.2 Lactation
Risk Summary
There is no information regarding the presence of evolocumab in human milk, the effects on the breastfed infant, or the effects on milk production. Human IgG is present in human milk, but published data suggest that breast milk antibodies do not enter the neonatal and infant circulation in substantial amounts.
The development and health benefits of breastfeeding should be considered along with the mother's clinical need for REPATHA and any potential adverse effects on the breastfed infant from REPATHA or from the underlying maternal condition.
8.4 Pediatric Use
The safety and effectiveness of REPATHA in combination with diet and other LDL-C-lowering therapies for the treatment of HoFH have been established in pediatric patients aged 10 years and older. Use of REPATHA for this indication is supported by evidence from an adequate and well-controlled trial in adults and pediatric patients aged 13 years and older with HoFH (including 7 pediatric patients treated with REPATHA) and from open-label studies which included an additional 19 pediatric patients aged 11 years and older with HoFH not previously treated with REPATHA [see Adverse Reactions (6.1) and Clinical Studies (14)].
The safety and effectiveness of REPATHA as an adjunct to diet and other LDL-C- lowering therapies for the treatment of HeFH have been established in pediatric patients aged 10 years and older. Use of REPATHA for this indication is based on data from a 24-week, randomized, placebo-controlled, double-blind trial in pediatric patients with HeFH. In the trial, 104 patients received REPATHA 420 mg subcutaneously once monthly and 53 patients received placebo; 39 patients (25%) were 10 to 11 years of age [see Adverse Reactions (6.1) and Clinical Studies (14)].
The safety and effectiveness of REPATHA have not been established in pediatric patients with HeFH or HoFH who are younger than 10 years old or in pediatric patients with other types of hyperlipidemia.
8.5 Geriatric Use
In controlled trials, 7656 (41%) patients treated with REPATHA were ≥ 65 years old and 1500 (8%) were ≥ 75 years old. No overall differences in safety or effectiveness were observed between these patients and younger patients, and other reported clinical experience has not identified differences in responses between the elderly and younger patients, but greater sensitivity of some older individuals cannot be ruled out.
8.6 Renal Impairment
No dose adjustment is needed in patients with renal impairment [see Clinical Pharmacology (12.3)].
8.7 Hepatic Impairment
No dose adjustment is needed in patients with mild to moderate hepatic impairment (Child-Pugh A or B). No data are available in patients with severe hepatic impairment [see Clinical Pharmacology (12.3)].
SPL PATIENT PACKAGE INSERT SECTION
|
Patient Information | |
|---|---|
|
This Patient Information has been approved by the U.S. Food and Drug
Administration. |
Revised: 11/2024 |
|
What is REPATHA? | |
|
REPATHA is an injectable prescription medicine used: | |
| |
|
It is not known if REPATHA is safe and effective in children with HeFH or HoFH who are younger than 10 years of age or in children with other types of hyperlipidemia. | |
|
Who should not use REPATHA? | |
|
Do not use REPATHA if you or your child are allergic to evolocumab or to any of the ingredients in REPATHA. See the end of this leaflet for a complete list of ingredients in REPATHA. | |
|
What should I tell my healthcare provider before using REPATHA? | |
|
Before you or your child start using REPATHA, tell your healthcare provider about all your medical conditions, including if you or your child: | |
| |
|
If you or your child are pregnant or breastfeed during REPATHA treatment, you are encouraged to call Amgen at 1-800-772-6436 (1-800-77-AMGEN) or visit https://wwwext.amgen.com/products/global-patient-safety/adverse-event- reporting to share information about the health of you and your baby or your child and your child's baby. | |
|
Tell your healthcare provider or pharmacist about any prescription and over- the-counter medicines, vitamins, or herbal supplements you or your child take. | |
|
How should I use REPATHA? | |
|
*See the detailed "Instructions for Use" that comes with this Patient Information about the right way to prepare and give REPATHA.
| |
|
If you or your child are not sure when to take REPATHA after a missed dose, ask your healthcare provider or pharmacist. | |
| |
|
What are the possible side effects of REPATHA? | |
|
REPATHA can cause serious side effects including: | |
|
*Serious Allergic Reactions. Some people taking REPATHA have had serious allergic reactions. Stop taking REPATHA and call your healthcare provider or seek emergency medical help right away if you or your child have any of these symptoms: * trouble breathing or swallowing * raised bumps (hives) * rash, or itching * swelling of the face, lips, tongue, throat or arms | |
|
The most common side effects of REPATHA include: runny nose, sore throat, symptoms of the common cold, flu or flu-like symptoms, back pain, high blood sugar levels (diabetes) and redness, pain, or bruising at the injection site. | |
|
Tell your healthcare provider if you or your child have any side effect that bothers you or that does not go away. | |
|
These are not all the possible side effects of REPATHA. Ask your healthcare provider or pharmacist for more information. | |
|
Call your healthcare provider for medical advice about side effects. You may report side effects to FDA at 1-800-FDA-1088. | |
|
How should I store REPATHA?
Keep REPATHA and all medicines out of the reach of children. | |
|
General information about the safe and effective use of REPATHA. | |
|
Medicines are sometimes prescribed for purposes other than those listed in a Patient Information leaflet.Do not use REPATHA for a condition for which it was not prescribed.Do not give REPATHA to other people, even if they have the same symptoms that you or your child have. It may harm them. | |
|
You can ask your pharmacist or healthcare provider for information about REPATHA that is written for healthcare professionals. | |
|
What are the ingredients in REPATHA? | |
| |
|
Manufactured by: Amgen Inc. One Amgen Center Drive, Thousand Oaks, California 91320-1799. | |
|
U.S. License Number 1080 | |
|
Patent: http://pat.amgen.com/repatha/ | |
|
© 2017-2021, 2024 Amgen Inc. All rights reserved. | |
|
For more information about REPATHA, go to www.REPATHA.com or call 1-844-REPATHA (1-844-737-2842). |
DOSAGE FORMS & STRENGTHS SECTION
3 DOSAGE FORMS AND STRENGTHS
REPATHA is a clear to opalescent, colorless to pale yellow solution available as follows:
- Injection: 140 mg/mL solution in a prefilled single-dose SureClick® autoinjector
- Injection: 140 mg/mL solution in a prefilled single-dose syringe
- Injection: 420 mg/3.5 mL solution in a single-dose Pushtronex® system (on-body infusor with prefilled cartridge)
- Injection: 140 mg/mL solution prefilled single-dose SureClick® autoinjector (3)
- Injection: 140 mg/mL solution prefilled single-dose syringe (3)
- Injection: 420 mg/3.5 mL solution single-dose Pushtronex® system (on-body infusor with prefilled cartridge) (3)
DESCRIPTION SECTION
11 DESCRIPTION
Evolocumab is a human monoclonal immunoglobulin G2 (IgG2) directed against human proprotein convertase subtilisin kexin type 9 (PCSK9). Evolocumab has an approximate molecular weight (MW) of 144 kDa and is produced in genetically engineered mammalian (Chinese hamster ovary) cells.
REPATHA is a sterile, preservative-free, clear to opalescent, colorless to pale yellow solution for subcutaneous use. Each 1 mL prefilled single-dose SureClick® autoinjector and prefilled single-dose syringe contains 140 mg evolocumab, acetate (1.2 mg), polysorbate 80 (0.1 mg), proline (25 mg) in Water for Injection, USP. Sodium hydroxide may be used to adjust to a pH of 5.0. Each single-dose Pushtronex® system (on-body infusor with prefilled cartridge) delivers a 3.5 mL solution containing 420 mg evolocumab, acetate (4.2 mg), polysorbate 80 (0.35 mg), proline (89 mg) in Water for Injection, USP. Sodium hydroxide may be used to adjust to a pH of 5.0.
CLINICAL STUDIES SECTION
14 CLINICAL STUDIES
Adult Patients with Established Cardiovascular Disease
Study 1 (FOURIER, NCT01764633) was a double-blind, randomized, placebo- controlled, event-driven trial in 27,564 (13,784 REPATHA, 13,780 placebo) adult patients with established cardiovascular disease and with LDL-C ≥ 70 mg/dL and/or non-HDL-C ≥ 100 mg/dL despite high- or moderate-intensity statin therapy. Patients were randomly assigned 1:1 to receive either subcutaneous injections of REPATHA (140 mg every 2 weeks or 420 mg once monthly) or placebo; 86% used the every-2-week regimen throughout the trial. The median follow-up duration was 26 months. Overall, 99.2% of patients were followed until the end of the trial or death.
The mean (SD) age at baseline was 63 (9) years, with 45% being at least 65 years old; 25% were women. The trial population was 85% White, 2% Black, and 10% Asian; 8% identified as Hispanic ethnicity. Regarding prior diagnoses of cardiovascular disease, 81% had prior myocardial infarction, 19% prior non- hemorrhagic stroke, and 13% had symptomatic peripheral arterial disease. Selected additional baseline risk factors included hypertension (80%), diabetes mellitus (1% type 1; 36% type 2), current daily cigarette smoking (28%), New York Heart Association class I or II congestive heart failure (23%), and eGFR < 60 mL/min per 1.73 m2 (6%). Most patients were on a high- (69%) or moderate-intensity (30%) statin therapy at baseline, and 5% were also taking ezetimibe. Most patients were taking at least one other cardiovascular medication including anti-platelet agents (93%), beta blockers (76%), angiotensin converting enzyme (ACE) inhibitors (56%), or angiotensin receptor blockers (23%). On stable background lipid-lowering therapy, the median [Q1, Q3] LDL-C at baseline was 92 [80, 109] mg/dL; the mean (SD) was 98 (28) mg/dL.
REPATHA significantly reduced the risk for the primary composite endpoint (time to first occurrence of cardiovascular death, myocardial infarction, stroke, hospitalization for unstable angina, or coronary revascularization; p < 0.0001) and the key secondary composite endpoint (time to first occurrence of cardiovascular death, myocardial infarction, or stroke; p < 0.0001). The Kaplan-Meier estimates of the cumulative incidence of the primary and key secondary composite endpoints over time are shown in Figure 1 and Figure 2 below.
The results of primary and secondary efficacy endpoints are shown in Table 3 below.
Table 3. Effect of REPATHA on Cardiovascular Events in Patients with Established Cardiovascular Disease in FOURIER|
Placebo |
REPATHA |
REPATHA vs. Placebo | |||
|---|---|---|---|---|---|
|
N = 13780 |
Incidence Rate (per 100 patient years) |
N = 13784 |
Incidence Rate (per 100 patient years) |
Hazard Ratio | |
| |||||
|
Primary composite endpoint | |||||
|
Time to first occurrence of cardiovascular death, myocardial infarction, stroke, coronary revascularization, hospitalization for unstable angina |
1563 (11.3) |
5.2 |
1344 (9.8) |
4.5 |
0.85 |
|
Key secondary composite endpoint | |||||
|
Time to first occurrence of cardiovascular death, myocardial infarction, stroke |
1013 (7.4) |
3.4 |
816 (5.9) |
2.7 |
0.80 |
|
Other secondary endpoints | |||||
|
Time to cardiovascular death |
240 (1.7) |
0.8 |
251 (1.8) |
0.8 |
1.05 |
|
Time to death by any cause* |
426 (3.1) |
1.4 |
444 (3.2) |
1.5 |
1.04 |
|
Time to first fatal or non-fatal myocardial infarction |
639 (4.6) |
2.1 |
468 (3.4) |
1.6 |
0.73 |
|
Time to first fatal or non-fatal stroke |
262 (1.9) |
0.9 |
207 (1.5) |
0.7 |
0.79 |
|
Time to first coronary revascularization |
965 (7.0) |
3.2 |
759 (5.5) |
2.5 |
0.78 |
|
Time to first hospitalization for unstable angina† |
239 (1.7) |
0.8 |
236 (1.7) |
0.8 |
0.99 |
Figure 1. Estimated Cumulative Incidence of Primary Composite Endpoint Over 3 Years in FOURIER
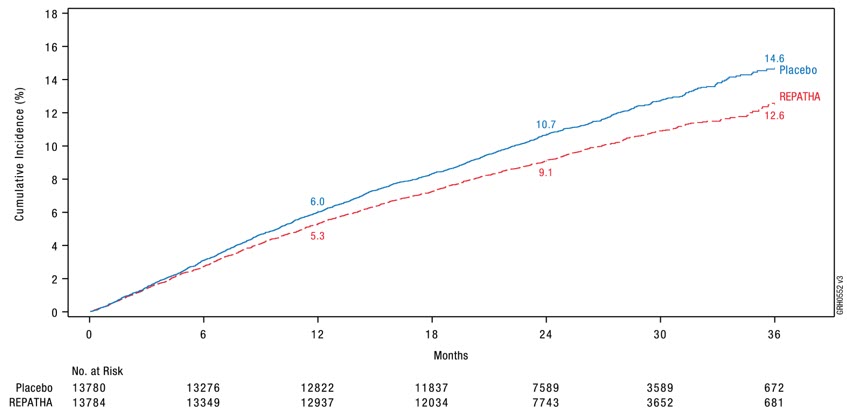
Figure 2. Estimated Cumulative Incidence of Key Secondary Composite Endpoint Over 3 Years in FOURIER
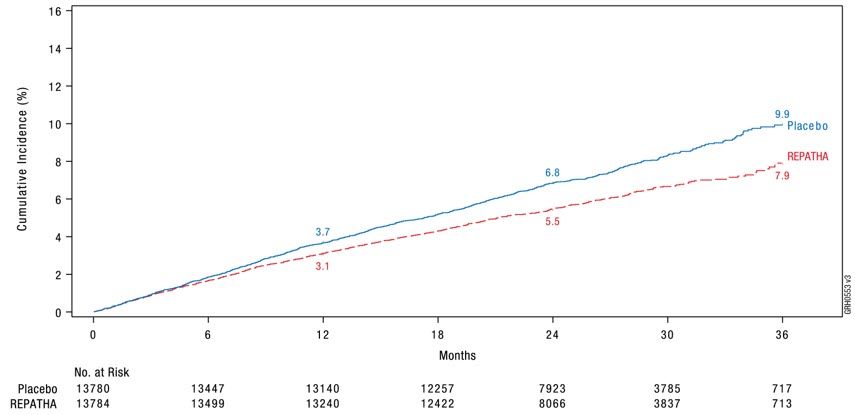
The difference between REPATHA and placebo in mean percent change in LDL-C from baseline to Week 12 was −63% (95% CI: −63%, −62%) and from baseline to Week 72 was −57% (95% CI: −58%, −56%). At Week 48, the median [Q1, Q3] LDL-C was 26 [15, 46] mg/dL in the REPATHA group, with 47% of patients having LDL-C < 25 mg/dL.
In EBBINGHAUS (NCT02207634), a substudy of 1974 patients enrolled in the FOURIER trial, REPATHA was non-inferior to placebo on selected cognitive function domains as assessed with the use of neuropsychological function tests over a median follow-up of 19 months.
Primary Hyperlipidemia
Study 2 (LAPLACE-2, NCT01763866) was a multicenter, double-blind, randomized controlled 12-week trial in which patients were initially randomized to an open-label specific statin regimen for a 4-week lipid stabilization period followed by random assignment to subcutaneous injections of REPATHA 140 mg every 2 weeks, REPATHA 420 mg once monthly, or placebo for 12 weeks. The trial included 1896 patients with hyperlipidemia who received REPATHA, placebo, or ezetimibe as add-on therapy to daily doses of statins (atorvastatin, rosuvastatin, or simvastatin). Ezetimibe was also included as an active control only among those assigned to background atorvastatin. Overall, the mean age at baseline was 60 years (range: 20 to 80 years), 35% were ≥ 65 years old, 46% women, 94% White, 4% were Black, and 1% Asian; 5% identified as Hispanic or Latino ethnicity. After 4 weeks of background statin therapy, the mean baseline LDL-C ranged between 77 and 127 mg/dL across the five background therapy arms.
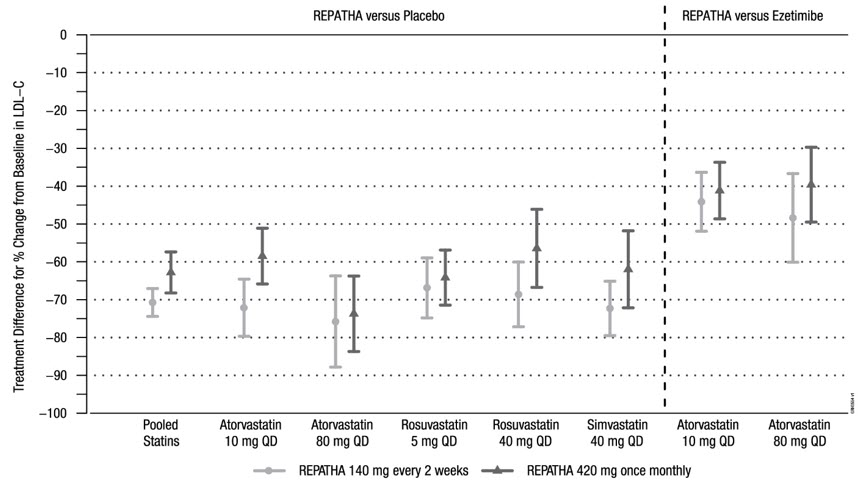
The difference between REPATHA and placebo in mean percent change in LDL-C from baseline to Week 12 was −71% (95% CI: −74%, −67%; p < 0.0001) and −63% (95% CI: −68%, −57%; p < 0.0001) for the 140 mg every 2 weeks and 420 mg once monthly dosages, respectively. The difference between REPATHA and ezetimibe in mean percent change in LDL-C from baseline to Week 12 was −45% (95% CI: −52%, −39%; p < 0.0001) and −41% (95% CI: −47%, −35%; p < 0.0001) for the 140 mg every 2 weeks and 420 mg once monthly dosages, respectively. For additional results, see Table 4 and Figure 3.
Table 4. Effect of REPATHA on Lipid Parameters in Patients with Hyperlipidemia on Background Statin Regimens (Mean % Change from Baseline to Week 12 in LAPLACE-2)|
Treatment Group |
LDL-C |
Non-HDL-C |
Apo B |
Total Cholesterol |
|---|---|---|---|---|
|
Estimates based on a multiple imputation model that accounts for treatment adherence | ||||
| ||||
|
REPATHA every 2 weeks vs. Placebo every 2 weeks | ||||
|
Placebo every 2 weeks (n = 281) |
8 |
6 |
5 |
4 |
|
REPATHA 140 mg every 2 weeks* (n = 555) |
-63 |
-53 |
-49 |
-36 |
|
Mean difference from placebo |
-71 |
-59 |
-55 |
-40 |
|
REPATHA once monthly vs. Placebo once monthly | ||||
|
Placebo once monthly (n = 277) |
4 |
5 |
3 |
2 |
|
REPATHA 420 mg once monthly (n = 562) |
-59 |
-50 |
-46 |
-34 |
|
Mean difference from placebo |
-63 |
-54 |
-50 |
-36 |
|
REPATHA every 2 weeks vs. Ezetimibe 10 mg daily | ||||
|
Ezetimibe 10 mg daily (n = 112) |
-17 |
-16 |
-14 |
-12 |
|
REPATHA 140 mg every 2 weeks* (n = 219) |
-63 |
-52 |
-49 |
-36 |
|
Mean difference from Ezetimibe |
-45 |
-36 |
-35 |
-24 |
|
REPATHA once monthly vs. Ezetimibe 10 mg daily | ||||
|
Ezetimibe 10 mg daily (n = 109) |
-19 |
-16 |
-11 |
-12 |
|
REPATHA 420 mg once monthly (n = 220) |
-59 |
-50 |
-46 |
-34 |
|
Mean difference from Ezetimibe |
-41 |
-35 |
-34 |
-22 |
Figure 3. Effect of REPATHA on LDL-C in Patients with Hyperlipidemia when Combined with Statins (Mean % Change from Baseline to Week 12 in LAPLACE-2)
|
Estimates based on a multiple imputation model that accounts for treatment
adherence |
|
|
Study 3 (DESCARTES, NCT01516879) was a multicenter, double-blind, randomized, placebo-controlled, 52-week trial that included 901 patients with hyperlipidemia who received protocol-determined background lipid-lowering therapy of a cholesterol-lowering diet either alone or in addition to atorvastatin (10 mg or 80 mg daily) or the combination of atorvastatin 80 mg daily with ezetimibe. After stabilization on background therapy, patients were randomly assigned to the addition of placebo or REPATHA 420 mg administered subcutaneously once monthly. Overall, the mean age at baseline was 56 years (range: 25 to 75 years), 23% were ≥ 65 years, 52% women, 80% White, 8% Black, and 6% Asian; 6% identified as Hispanic or Latino ethnicity. After stabilization on the assigned background therapy, the mean baseline LDL-C ranged between 90 and 117 mg/dL across the four background therapy groups.
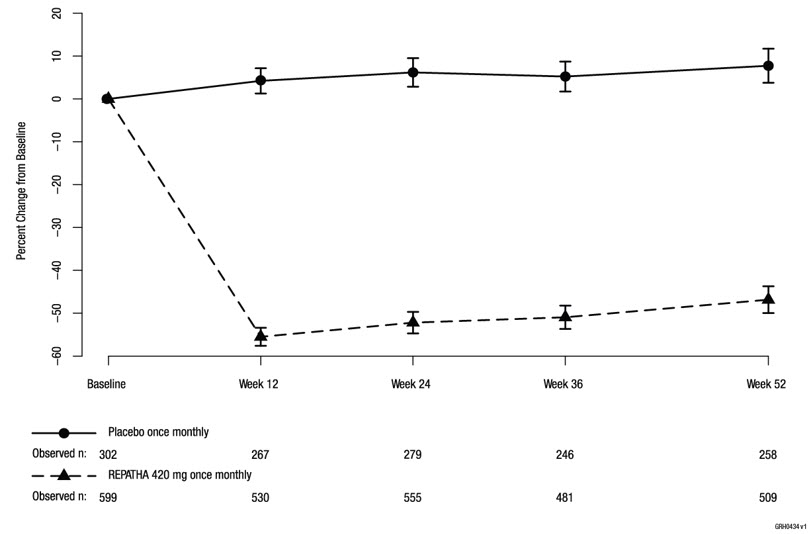
In these patients with hyperlipidemia on a protocol-determined background therapy, the difference between REPATHA 420 mg once monthly and placebo in mean percent change in LDL-C from baseline to Week 52 was −55% (95% CI: −60%, −50%; p < 0.0001) (Table 5 and Figure 4). For additional results, see Table 5.
Table 5. Effect of REPATHA on Lipid Parameters in Patients with Hyperlipidemia* (Mean % Change from Baseline to Week 52 in DESCARTES)|
Treatment Group |
LDL-C |
Non-HDL-C |
Apo B |
Total Cholesterol |
|---|---|---|---|---|
|
Estimates based on a multiple imputation model that accounts for treatment adherence | ||||
| ||||
|
Placebo once monthly (n = 302) |
8 |
8 |
2 |
5 |
|
REPATHA 420 mg once monthly (n = 599) |
-47 |
-39 |
-38 |
-26 |
|
Mean difference from placebo |
-55 |
-46 |
-40 |
-31 |
Figure 4. Effect of REPATHA 420 mg Once Monthly on LDL-C in Patients with Hyperlipidemia in DESCARTES
|
Estimates based on a multiple imputation model that accounts for treatment
adherence |
|
|
Study 4 (MENDEL-2, NCT01763827) was a multicenter, double-blind, randomized, placebo- and active-controlled, 12-week trial that included 614 patients with hyperlipidemia who were not taking lipid-lowering therapy at baseline. Patients were randomly assigned to receive subcutaneous injections of REPATHA 140 mg every 2 weeks, REPATHA 420 mg once monthly, or placebo for 12 weeks. Blinded administration of ezetimibe was also included as an active control. Overall, the mean age at baseline was 53 years (range: 20 to 80 years), 18% were ≥ 65 years old, 66% were women, 83% White, 7% Black, and 9% Asian; 11% identified as Hispanic or Latino ethnicity. The mean baseline LDL-C was 143 mg/dL.
The difference between REPATHA and placebo in mean percent change in LDL-C from baseline to Week 12 was −55% (95% CI: −60%, −50%; p < 0.0001) and −57% (95% CI: −61%, −52%; p < 0.0001) for the 140 mg every 2 weeks and 420 mg once monthly dosages, respectively. The difference between REPATHA and ezetimibe in mean percent change in LDL-C from baseline to Week 12 was −37% (95% CI: −42%, −32%; p < 0.0001) and −38% (95% CI: −42%, −34%; p < 0.0001) for the 140 mg every 2 weeks and 420 mg once monthly dosages, respectively. For additional results, see Table 6.
Table 6. Effect of REPATHA on Lipid Parameters in Patients with Hyperlipidemia (Mean % Change from Baseline to Week 12 in MENDEL-2)|
Treatment Group |
LDL-C |
Non-HDL-C |
Apo B |
Total Cholesterol |
|---|---|---|---|---|
|
Estimates based on a multiple imputation model that accounts for treatment adherence | ||||
| ||||
|
Placebo every 2 weeks (n = 76) |
1 |
0 |
1 |
0 |
|
Ezetimibe 10 mg daily (n = 77) |
-17 |
-14 |
-13 |
-10 |
|
REPATHA 140 mg every 2 weeks* (n = 153) |
-54 |
-47 |
-44 |
-34 |
|
Mean difference from placebo |
-55 |
-47 |
-45 |
-34 |
|
Mean difference from Ezetimibe |
-37 |
-33 |
-32 |
-23 |
|
Placebo once monthly (n = 78) |
1 |
2 |
2 |
0 |
|
Ezetimibe 10 mg daily (n = 77) |
-18 |
-16 |
-13 |
-12 |
|
REPATHA 420 mg once monthly (n = 153) |
-56 |
-49 |
-46 |
-35 |
|
Mean difference from placebo |
-57 |
-51 |
-48 |
-35 |
|
Mean difference from Ezetimibe |
-38 |
-32 |
-33 |
-23 |
Study 5 (RUTHERFORD-2, NCT01763918) was a multicenter, double-blind, randomized, placebo-controlled, 12-week trial in 329 patients with HeFH on statins with or without other lipid-lowering therapies. Patients were randomized to receive subcutaneous injections of REPATHA 140 mg every two weeks, 420 mg once monthly, or placebo. HeFH was diagnosed by the Simon Broome criteria (1991). In Study 5, 38% of patients had clinical atherosclerotic cardiovascular disease. The mean age at baseline was 51 years (range: 19 to 79 years), 15% of the patients were ≥ 65 years old, 42% were women, 90% were White, 5% were Asian, and 1% were Black. The average LDL-C at baseline was 156 mg/dL with 76% of the patients on high-intensity statin therapy.
The differences between REPATHA and placebo in mean percent change in LDL-C from baseline to Week 12 was −61% (95% CI: −67%, −55%; p < 0.0001) and −60% (95% CI: −68%, −52%; p < 0.0001) for the 140 mg every 2 weeks and 420 mg once monthly dosages, respectively. For additional results, see Table 7 and Figure 5.
Table 7. Effect of REPATHA on Lipid Parameters in Patients with HeFH (Mean % Change from Baseline to Week 12 in RUTHERFORD-2)|
Treatment Group |
LDL-C |
Non-HDL-C |
Apo B |
Total Cholesterol |
|---|---|---|---|---|
|
Estimates based on a multiple imputation model that accounts for treatment adherence | ||||
| ||||
|
Placebo every 2 weeks (n = 54) |
-1 |
-1 |
-1 |
-2 |
|
REPATHA 140 mg every 2 weeks* (n = 110) |
-62 |
-56 |
-49 |
-42 |
|
Mean difference from placebo |
-61 |
-54 |
-49 |
-40 |
|
Placebo once monthly (n = 55) |
4 |
4 |
4 |
2 |
|
REPATHA 420 mg once monthly (n = 110) |
-56 |
-49 |
-44 |
-37 |
|
Mean difference from placebo |
-60 |
-53 |
-48 |
-39 |
Figure 5. Effect of REPATHA on LDL-C in Patients with HeFH (Mean % Change from Baseline to Week 12 in RUTHERFORD-2)
|
N = number of patients randomized and dosed in the full analysis set |
|
|
Pediatric Patients with HeFH
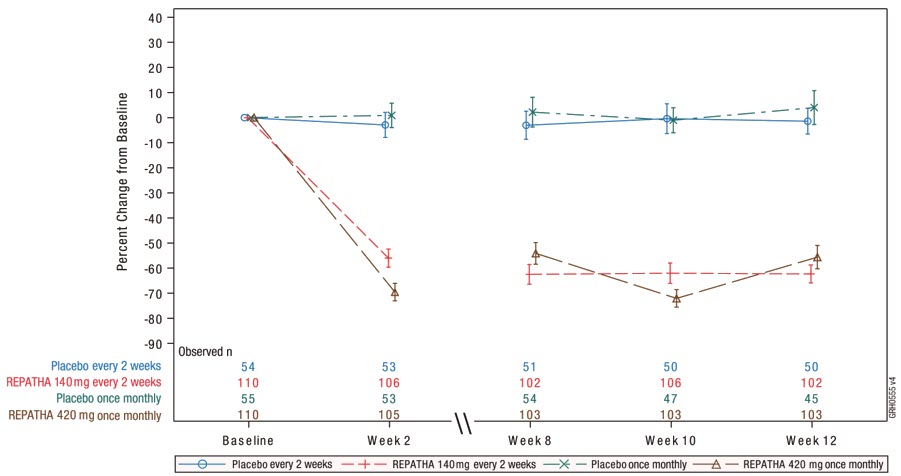
Study 6 (HAUSER-RCT, NCT02392559) was a randomized, multicenter, placebo- controlled, double-blind, 24-week trial in 157 pediatric patients aged 10 to 17 years with HeFH [see Use in Specific Populations (8.4)]. HeFH was diagnosed by diagnostic criteria for HeFH [Simon Broome Register Group (1991), the Dutch Lipid Clinic Network (1999), MEDPED (1993)] or by genetic testing. Patients were required to be on a low-fat diet and optimized background lipid-lowering therapy. Patients were randomly assigned 2:1 to receive 24 weeks of subcutaneous once monthly 420 mg REPATHA or placebo; 104 patients received REPATHA and 53 patients received placebo. The mean age was 14 years (range: 10 to 17 years), 56% were female, 85% White, 1% Black, 1% Asian, 13% Other, and 8% Hispanic. The mean LDL-C at baseline was 184 mg/dL; 17% of patients were on high-intensity statin, 62% on moderate-intensity statin, and 13% on ezetimibe.
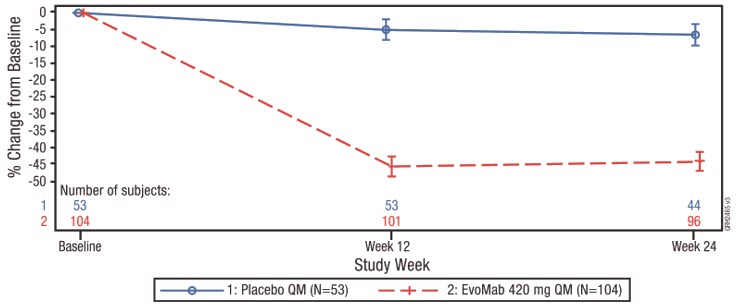
The difference between REPATHA and placebo in mean percent change in LDL-C from baseline to Week 24 was −38% (95% CI: −45%, −31%; p < 0.0001). For additional results, see Table 8 and Figure 6.
Figure 6. Effect of REPATHA on LDL-C in Pediatric Patients with HeFH (Mean % Change from Baseline in HAUSER-RCT)
|
EvoMab = evolocumab; LDL-C = low density lipoprotein cholesterol; QM = monthly
(subcutaneous) |
|
|
|
Treatment Group |
LDL-C |
Non-HDL-C |
Apo B |
Total Cholesterol |
|---|---|---|---|---|
|
All adjusted p-values < 0.0001. | ||||
|
Placebo once monthly |
-6 |
-6 |
-2 |
-5 |
|
REPATHA 420 mg once monthly |
-44 |
-41 |
-35 |
-32 |
|
Mean difference from placebo |
-38 |
-35 |
-32 |
-27 |
Adults and Pediatric Patients with HoFH
Study 7 (TESLA, NCT01588496) was a multicenter, double-blind, randomized, placebo-controlled, 12-week trial in 49 patients (not on lipid-apheresis therapy) with HoFH. In this trial, 33 patients received subcutaneous injections of 420 mg of REPATHA once monthly and 16 patients received placebo as an adjunct to other lipid-lowering therapies (e.g., statins, ezetimibe). The mean age at baseline was 31 years, 49% were women, 90% White, 4% were Asian, and 6% other. The trial included 10 adolescents (ages 13 to 17 years), 7 of whom received REPATHA. The mean LDL-C at baseline was 349 mg/dL with all patients on statins (atorvastatin or rosuvastatin) and 92% on ezetimibe. The diagnosis of HoFH was made by genetic confirmation or a clinical diagnosis based on a history of an untreated LDL-C concentration > 500 mg/dL together with either xanthoma before 10 years of age or evidence of HeFH in both parents.
The difference between REPATHA and placebo in mean percent change in LDL-C from baseline to Week 12 was −31% (95% CI: −44%, −18%; p < 0.0001). For additional results, see Table 9.
Patients known to have two LDL-receptor negative alleles (little to no residual function) did not respond to REPATHA.
Table 9. Effect of REPATHA on Lipid Parameters in Patients with HoFH (Mean % Change from Baseline to Week 12 in TESLA)|
Treatment Group |
LDL-C |
Non-HDL-C |
Apo B |
Total Cholesterol |
|---|---|---|---|---|
|
Estimates based on a multiple imputation model that accounts for treatment adherence | ||||
|
Placebo once monthly (n = 16) |
9 |
8 |
4 |
8 |
|
REPATHA 420 mg once monthly (n = 33) |
-22 |
-20 |
-17 |
-17 |
|
Mean difference from placebo |
-31 |
-28 |
-21 |
-25 |
Study 8 (TAUSSIG, NCT01624142) was a multicenter, open-label 5-year extension study with REPATHA in 106 patients with HoFH, who were treated with REPATHA as an adjunct to other lipid-lowering therapies. The study included 14 pediatric patients (ages 13 to 17 years). All patients in the study were initially treated with REPATHA 420 mg once monthly except for those receiving lipid apheresis at enrollment, who began with REPATHA 420 mg every 2 weeks. Dose frequency in non-apheresis patients could be titrated up to 420 mg once every 2 weeks based on LDL-C response and PCSK9 levels.
A total of 48 patients with HoFH received REPATHA 420 mg once monthly for at least 12 weeks in Study 8 followed by REPATHA 420 mg every 2 weeks for at least 12 weeks. Mean percent change from baseline in LDL-C were −20% at Week 12 of 420 mg once monthly treatment and −30% at Week 12 of 420 mg every 2 weeks treatment, based on available data.
Study 9 (HAUSER-OLE, NCT02624869) was an open-label, single-arm, multicenter, 80-week study to evaluate the safety, tolerability, and efficacy of REPATHA for LDL-C reduction in pediatric patients aged 10 to 17 years with HoFH [see Use in Specific Populations (8.4)]. Patients were on a low-fat diet and receiving background lipid-lowering therapy. Overall, 12 patients with HoFH received 420 mg REPATHA subcutaneously once monthly. The mean age was 12 years (range 11 to 17 years), 17% were female, 75% White, 17% Asian, and 8% Other. Median (Q1, Q3) LDL-C at baseline was 398 (343, 475) mg/dL, and all patients were on statins (atorvastatin or rosuvastatin) and ezetimibe. No patients were receiving lipid apheresis. The diagnosis of HoFH was made by genetic confirmation in all patients but enrollment by a clinical diagnosis was permitted. The median (Q1, Q3) percent change in LDL-C from baseline to Week 80 was −14% (−41, 4). Two of the 3 subjects with < 5% LDLR activity responded to evolocumab treatment.
HOW SUPPLIED SECTION
16 HOW SUPPLIED/STORAGE AND HANDLING
REPATHA is a clear to opalescent, colorless to pale yellow solution supplied as follows:
Not Made with Natural Rubber Latex –|
140 mg/mL prefilled single-dose SureClick® autoinjector |
2 pack |
NDC 72511-393-02 |
|
140 mg/mL prefilled single-dose SureClick® autoinjector |
1 pack |
NDC 72511-393-01 |
|
140 mg/mL prefilled single-dose Syringe |
1 pack |
NDC 72511-501-01 |
|
420 mg/3.5 mL single-dose Pushtronex® system (on-body infusor with prefilled cartridge) |
1 pack |
NDC 72511-770-01 |
| ||
|
140 mg/mL prefilled single-dose SureClick® autoinjector* |
2 pack |
NDC 72511-760-02 |
|
140 mg/mL prefilled single-dose Syringe* |
1 pack |
NDC 72511-750-01 |
Store refrigerated at 2°C to 8°C (36°F to 46°F) in the original carton to protect from light. Do not freeze. Do not shake.
For convenience, REPATHA may be kept at room temperature at 68°F to 77°F (20°C to 25°C) in the original carton for 30 days. If not used within the 30 days, discard REPATHA.
INFORMATION FOR PATIENTS SECTION
17 PATIENT COUNSELING INFORMATION
Advise the patient and/or caregiver to read the FDA-Approved Patient Labeling (Patient Information and Instructions for Use).
Hypersensitivity
Inform patients that serious hypersensitivity reactions (e.g., angioedema) have been reported in patients treated with REPATHA. Advise patients on the symptoms of hypersensitivity reactions and instruct them to discontinue REPATHA and seek medical attention promptly, if such symptoms occur.
Latex-Sensitivity
Instruct patients to inform their healthcare provider if they are sensitive to latex. Inform patients that REPATHA is available as prefilled single-dose SureClick® autoinjectors and prefilled single-dose syringes that either contain dry natural rubber (a derivative of latex) in the needle cover or are not made with natural rubber latex, and the carton and Instructions for Use state if the product contains dry natural rubber. Advise latex-sensitive patients that the needle cover of the glass prefilled single-dose SureClick® autoinjector and prefilled single-dose syringe that contain dry natural rubber (a derivative of latex) may cause allergic reactions in individuals sensitive to latex. [see How Supplied/Storage and Handling (16)].
Pregnancy
Advise women who are exposed to REPATHA during pregnancy that there is a pregnancy safety study that monitors pregnancy outcomes. Encourage these patients to report their pregnancy to Amgen at 1-800-77-AMGEN (1-800-772-6436) or https://wwwext.amgen.com/products/global-patient-safety/adverse-event- reporting [see Use in Specific Populations (8.1)].
Administration
Provide guidance to patients and caregivers on proper subcutaneous administration technique and how to use the prefilled single-dose SureClick® autoinjector, prefilled single-dose syringe, or single-dose on-body infusor with prefilled cartridge correctly. Inform patients that it may take up to 15 seconds to administer REPATHA using the prefilled single-dose SureClick® autoinjector or prefilled single-dose syringe and about 5 minutes to administer REPATHA using the single-dose on-body infusor with prefilled cartridge.
The single-dose on-body infusor with prefilled cartridge is not made with natural rubber latex.
SPL UNCLASSIFIED SECTION
REFERENCE GUIDE
|
Side 1 |
Reference Guide |
Read all instructions in carton before use. Questions? Call 1-844-REPATHA |
|
Guide to parts |
Wait at least 30 minutes for the autoinjector to come to room temperature before giving your injection. |
Pull the orange cap straight off only when you are ready to inject. | |||
|
| |||||
|
2 | |||||
|
| |||||
|
Prepare and clean your injection site. | |||||
|
1 |
| ||||
|
Turn over to continue |
|
Side 2 |
Reference Guide |
AMGEN® |
Read other side first | |||||
|
Manufactured by:
| ||||||||
|
Stretchor pinch your injection site to create a firm surface. |
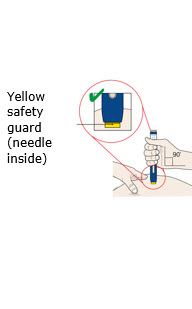
Put the yellow safety guard (needle inside) on your skin at 90 degrees. |
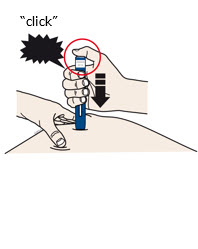
Firmlypush down the autoinjector onto your skin until it stops moving. |
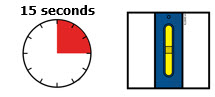
Keeppushing down on your skin forabout 15 seconds. | |||||
|
3 |
4 |
5 |
6 | |||||
|
|
|
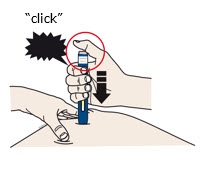
When you are ready to inject,press the gray start button. You will hear a
click. |
| |||||
|
Window turns from clear to yellow when the injection is done. |
 This
printed material is recyclable.
This
printed material is recyclable.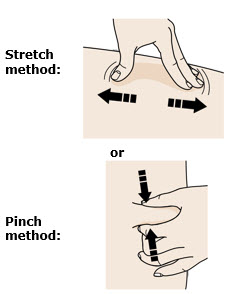
DOSAGE & ADMINISTRATION SECTION
2 DOSAGE AND ADMINISTRATION
2.1 Recommended Dosage
- In adults with established cardiovascular disease or with primary hyperlipidemia:
- The recommended dosage of REPATHA is either 140 mg every 2 weeks OR 420 mg once monthly administered subcutaneously [see Dosage and Administration (2.3)].
- If switching dosage regimens, administer the first dose of the new regimen on the next scheduled date of the prior regimen.
- In pediatric patients aged 10 years and older with HeFH:
- The recommended dosage of REPATHA is either 140 mg every 2 weeks OR 420 mg once monthly administered subcutaneously [see Dosage and Administration (2.3)].
- If switching dosage regimens, administer the first dose of the new regimen on the next scheduled date of the prior regimen.
- In adults and pediatric patients aged 10 years and older with HoFH:
- The initial recommended dosage of REPATHA is 420 mg once monthly administered subcutaneously [see Dosage and Administration (2.3)].
- The dosage can be increased to 420 mg every 2 weeks if a clinically meaningful response is not achieved in 12 weeks.
- Patients on lipid apheresis may initiate treatment with 420 mg every 2 weeks to correspond with their apheresis schedule. Administer REPATHA after the apheresis session is complete.
- Assess LDL-C when clinically appropriate. The LDL-lowering effect of REPATHA may be measured as early as 4 weeks after initiation.
- When monitoring LDL-C for patients receiving REPATHA 420 mg once monthly, note that LDL-C can vary during the dosing interval in some patients; recommend measuring LDL-C just prior to the next scheduled dose [see Clinical Studies (14)].
2.2 Missed Doses
If a dose is missed:
- Within 7 days from the missed dose, instruct the patient to administer REPATHA and resume the patient's original schedule.
- More than 7 days after the missed dose:
- For an every 2-week dose, instruct the patient to wait until the next dose on the original schedule.
- For a once-monthly dose, instruct the patient to administer the dose and start a new schedule based on this date.
2.3 Important Administration Instructions
- REPATHA is available as prefilled single-dose SureClick® autoinjectors and prefilled single-dose syringes that either contain dry natural rubber (a derivative of latex) in the needle cover or are not made with natural rubber latex [see How Supplied/Storage and Handling (16)]. Consider prescribing a presentation of REPATHA that does not contain dry natural rubber for individuals that are sensitive to latex [see Warnings and Precautions (5.1)].
- Train patients and/or caregivers on how to prepare and administer REPATHA, according to the Instructions for Use and instruct them to read and follow the Instructions for Use each time they use REPATHA.
- Prior to use, allow REPATHA to warm to room temperature for at least 30 minutes for the prefilled single-dose SureClick® autoinjector or prefilled single-dose syringe and for at least 45 minutes for the on-body infusor with prefilled cartridge if REPATHA has been refrigerated [see How Supplied/Storage and Handling (16)].
- Visually inspect REPATHA prior to administration. REPATHA is a clear to opalescent, colorless to pale yellow solution. Do not use if the solution is cloudy, discolored, or contains particles.
- Administer REPATHA subcutaneously into areas of the abdomen, thigh, or upper arm that are not tender, bruised, red, or indurated. Avoid injecting into areas with scars or stretch marks. Rotate injection sites for each administration.
- The 420 mg dose of REPATHA can be administered:
- over 5 minutes by using the single-dose on-body infusor with prefilled cartridge, or
- by giving 3 injections consecutively within 30 minutes using the prefilled single-dose SureClick® autoinjector or prefilled single-dose syringe.
In adults with established CVD or with primary hyperlipidemia:
- The recommended dosage of REPATHA is either 140 mg every 2 weeks OR 420 mg once monthly administered subcutaneously. (2.1)
- If switching dosage regimens, administer the first dose of the new regimen on the next scheduled date of the prior regimen. (2.1)
In pediatric patients aged 10 years and older with HeFH:
- The recommended dosage of REPATHA is either 140 mg every 2 weeks OR 420 mg once monthly administered subcutaneously. (2.1)
- If switching dosage regimens, administer the first dose of the new regimen on the next scheduled date of the prior regimen. (2.1)
In adults and pediatric patients aged 10 years and older with HoFH:
-
The initial recommended dosage of REPATHA is 420 mg once monthly administered subcutaneously. (2.1)
-
The dosage can be increased to 420 mg every 2 weeks if a clinically meaningful response is not achieved in 12 weeks. (2.1)
-
Patients on lipid apheresis may initiate treatment with 420 mg every 2 weeks to correspond with their apheresis schedule. Administer REPATHA after the apheresis session is complete. (2.1)
-
Assess LDL-C when clinically appropriate. The LDL-lowering effect of REPATHA may be measured as early as 4 weeks after initiation. (2.1)
-
REPATHA is available as prefilled single-dose SureClick® autoinjectors and prefilled single-dose syringes that either contain dry natural rubber (a derivative of latex) in the needle cover or are not made with natural rubber latex. Consider prescribing a presentation of REPATHA that does not contain dry natural rubber for individuals that are sensitive to latex. (2.3, 16)
-
Administer REPATHA subcutaneously into areas of the abdomen, thigh, or upper arm. Rotate injection sites for each administration. (2.3)
-
See Full Prescribing Information for important administration instructions. (2.3)
RECENT MAJOR CHANGES SECTION
RECENT MAJOR CHANGES
|
Indications and Usage (1) |
11/2024 |
|
Dosage and Administration (2.3) |
11/2024 |
|
Warnings and Precautions (5.1) |
11/2024 |
CLINICAL PHARMACOLOGY SECTION
12 CLINICAL PHARMACOLOGY
12.1 Mechanism of Action
Evolocumab is a human monoclonal IgG2 directed against human proprotein convertase subtilisin kexin type 9 (PCSK9). PCSK9 binds to the low-density lipoprotein receptor (LDLR) on the surface of hepatocytes to promote LDLR degradation within the liver. By inhibiting the binding of PCSK9 to LDLR, evolocumab increases the number of LDLRs available to clear LDL from the blood, thereby lowering LDL-C levels.
12.2 Pharmacodynamics
Following single subcutaneous administration of 140 mg or 420 mg of evolocumab, maximum suppression of circulating unbound PCSK9 occurred by 4 hours. Unbound PCSK9 concentrations returned toward baseline when evolocumab concentrations decreased below the limit of quantitation. Maximum LDL-C reduction occurred by 2 weeks after a single-dose of 140 mg of evolocumab and by 3 weeks after a single-dose of 420 mg of evolocumab.
12.3 Pharmacokinetics
Evolocumab exhibits non-linear kinetics as a result of binding to PCSK9. Administration of the 140 mg dose in healthy volunteers resulted in a Cmax mean of 18.6 μg/mL and AUClast mean of 188 day∙μg/mL. Administration of the 420 mg dose in healthy volunteers resulted in a Cmax mean of 59.0 μg/mL and AUClast mean of 924 day∙μg/mL. Following a single 420 mg intravenous dose, the mean systemic clearance was estimated to be 12 mL/hr. An approximate 2- to 3-fold accumulation was observed in trough serum concentrations (Cmin 7.21) following 140 mg doses administered subcutaneously every 2 weeks or following 420 mg doses administered subcutaneously monthly (Cmin 11.2), and serum trough concentrations approached steady-state by 12 weeks of dosing.
Absorption
Following a single subcutaneous dose of 140 mg or 420 mg evolocumab administered to healthy adults, median peak serum concentrations were attained in 3 to 4 days, and estimated absolute bioavailability was 72%.
Distribution
Following a single 420 mg intravenous dose, the mean steady-state volume of distribution was estimated to be 3.3 L.
Elimination
Two elimination phases were observed for REPATHA. At low concentrations, the elimination is predominately through saturable binding to target (PCSK9), while at higher concentrations the elimination of REPATHA is largely through a non-saturable proteolytic pathway. REPATHA was estimated to have an effective half-life of 11 to 17 days.
Specific Populations
The pharmacokinetics of evolocumab were not affected by age, gender, race, or creatinine clearance across all approved populations [see Use in Specific Populations (8.5)].
The exposure of evolocumab decreased with increasing body weight. These differences are not clinically meaningful.
Pediatric Patients
The pharmacokinetics of REPATHA were evaluated in 103 pediatric patients aged 10 to 17 years with HeFH (Study 6) [see Use in Specific Populations (8.4), and Clinical Studies (14)]. Following subcutaneous administration of 420 mg REPATHA once monthly, mean trough serum concentrations were 22.4 mcg/mL and 25.8 mcg/mL over the Week 12 and Week 24 time points, respectively. The pharmacokinetics of REPATHA were evaluated in 12 pediatric patients aged 11 to 17 years with HoFH (Study 9) [see Use in Specific Populations (8.4), and Clinical Studies (14)]. Following subcutaneous administration of 420 mg REPATHA once monthly, mean serum trough concentrations were 20.3 mcg/mL and 17.6 mcg/mL at Week 12 and Week 80, respectively.
Renal Impairment
Since monoclonal antibodies are not known to be eliminated via renal pathways, renal function is not expected to impact the pharmacokinetics of evolocumab.
In a clinical trial of 18 patients with either normal renal function (estimated glomerular filtration rate [eGFR] ≥ 90 mL/min/1.73 m2, n = 6), severe renal impairment (eGFR < 30 mL/min/1.73 m2, n = 6), or end-stage renal disease (ESRD) receiving hemodialysis (n = 6), exposure to evolocumab after a single 140 mg subcutaneous dose was decreased in patients with severe renal impairment or ESRD receiving hemodialysis. Reductions in PCSK9 levels in patients with severe renal impairment or ESRD receiving hemodialysis was similar to those with normal renal function [see Use in Specific Populations (8.6)].
Hepatic Impairment
Following a single 140 mg subcutaneous dose of evolocumab in patients with mild or moderate hepatic impairment, a 20-30% lower mean Cmax and 40-50% lower mean AUC were observed as compared to healthy patients [see Use in Specific Populations (8.7)].
Drug Interaction Studies
An approximately 20% decrease in the Cmax and AUC of evolocumab was observed in adult patients co-administered with a high-intensity statin regimen. This difference is not clinically meaningful.
NONCLINICAL TOXICOLOGY SECTION
13 NONCLINICAL TOXICOLOGY
13.1 Carcinogenesis, Mutagenesis, Impairment of Fertility
The carcinogenic potential of evolocumab was evaluated in a lifetime study conducted in the hamster at dose levels of 10, 30, and 100 mg/kg administered every 2 weeks. There were no evolocumab-related tumors at the highest dose at systemic exposures up to 38- and 15-fold the recommended human doses of 140 mg every 2 weeks and 420 mg once monthly, respectively, based on plasma AUC. The mutagenic potential of evolocumab has not been evaluated; however, monoclonal antibodies are not expected to alter DNA or chromosomes.
There were no adverse effects on fertility (including estrous cycling, sperm analysis, mating performance, and embryonic development) at the highest dose in a fertility and early embryonic developmental toxicology study in hamsters when evolocumab was subcutaneously administered at 10, 30, and 100 mg/kg every 2 weeks. The highest dose tested corresponds to systemic exposures up to 30- and 12-fold the recommended human doses of 140 mg every 2 weeks and 420 mg once monthly, respectively, based on plasma AUC. In addition, there were no adverse evolocumab-related effects on surrogate markers of fertility (reproductive organ histopathology, menstrual cycling, or sperm parameters) in a 6-month chronic toxicology study in sexually mature monkeys subcutaneously administered evolocumab at 3, 30, and 300 mg/kg once weekly. The highest dose tested corresponds to 744- and 300-fold the recommended human doses of 140 mg every 2 weeks and 420 mg once monthly, respectively, based on plasma AUC.
13.2 Animal Toxicology and/or Pharmacology
During a 3-month toxicology study of 10 and 100 mg/kg once every 2 weeks evolocumab in combination with 5 mg/kg once daily rosuvastatin in adult monkeys, there were no effects of evolocumab on the humoral immune response to keyhole limpet hemocyanin (KLH) after 1 to 2 months exposure. The highest dose tested corresponds to exposures 54- and 21-fold higher than the recommended human doses of 140 mg every 2 weeks and 420 mg once monthly, respectively, based on plasma AUC. Similarly, there were no effects of evolocumab on the humoral immune response to KLH (after 3 to 4 months exposure) in a 6-month study in cynomolgus monkeys at dose levels up to 300 mg/kg once weekly evolocumab corresponding to exposures 744- and 300-fold greater than the recommended human doses of 140 mg every 2 weeks and 420 mg once monthly, respectively, based on plasma AUC.