
Pirfenidone
These highlights do not include all the information needed to use PIRFENIDONE TABLETS safely and effectively. See full prescribing information for PIRFENIDONE TABLETS. PIRFENIDONE tablets, for oral useInitial U.S. Approval: 2014
7f5f7cc9-394e-4da0-87eb-84cfff9707a3
HUMAN PRESCRIPTION DRUG LABEL
Apr 30, 2023
Sandoz Inc
DUNS: 005387188
Products 2
Detailed information about drug products covered under this FDA approval, including NDC codes, dosage forms, ingredients, and administration routes.
Pirfenidone
Product Details
FDA regulatory identification and product classification information
FDA Identifiers
Product Classification
Product Specifications
INGREDIENTS (13)
Pirfenidone
Product Details
FDA regulatory identification and product classification information
FDA Identifiers
Product Classification
Product Specifications
INGREDIENTS (11)
Drug Labeling Information
WARNINGS AND PRECAUTIONS SECTION
5 WARNINGS AND PRECAUTIONS
5.1 Elevated Liver Enzymes and Drug-Induced Liver Injury
Cases of drug-induced liver injury (DILI) have been observed with pirfenidone. In the postmarketing period, non-serious and serious cases of DILI, including severe liver injury with fatal outcome, have been reported. Patients treated with pirfenidone 2403 mg/day in three Phase 3 trials had a higher incidence of elevations in ALT or AST ≥3 × ULN than placebo patients (3.7% vs. 0.8%, respectively). Elevations ≥10 × ULN in ALT or AST occurred in 0.3% of patients in the pirfenidone 2403 mg/day group and in 0.2% of patients in the placebo group. Increases in ALT and AST ≥3 × ULN were reversible with dose modification or treatment discontinuation.
Conduct liver function tests (ALT, AST, and bilirubin) prior to the initiation of therapy with pirfenidone, monthly for the first 6 months, every 3 months thereafter, and as clinically indicated. Measure liver function tests promptly in patients who report symptoms that may indicate liver injury, including fatigue, anorexia, right upper abdominal discomfort, dark urine, or jaundice. Dosage modification or interruption may be necessary for liver enzyme elevations [see Dosage and Administration (2.1, 2.3)].
5.2 Photosensitivity Reaction or Rash
Patients treated with pirfenidone 2403 mg/day in the three Phase 3 studies had a higher incidence of photosensitivity reactions (9%) compared with patients treated with placebo (1%). The majority of the photosensitivity reactions occurred during the initial 6 months. Instruct patients to avoid or minimize exposure to sunlight (including sunlamps), to use a sunblock (SPF 50 or higher), and to wear clothing that protects against sun exposure. Additionally, instruct patients to avoid concomitant medications known to cause photosensitivity. Dosage reduction or discontinuation may be necessary in some cases of photosensitivity reaction or rash [see Dosage and Administration (2.3)].
5.3 Severe Cutaneous Adverse Reactions
Severe cutaneous adverse reactions (SCAR), including Stevens-Johnson syndrome (SJS), toxic epidermal necrolysis (TEN), and drug reaction with eosinophilia and systemic symptoms (DRESS), have been reported in association with the use of pirfenidone in the post-marketing setting. If signs or symptoms of SCAR occur, interrupt pirfenidone treatment until the etiology of the reaction has been determined. Consultation with a dermatologist is recommended. If a SCAR is confirmed, permanently discontinue pirfenidone.
5.4 Gastrointestinal Disorders
In the clinical studies, gastrointestinal events of nausea, diarrhea, dyspepsia, vomiting, gastro-esophageal reflux disease, and abdominal pain were more frequently reported by patients in the pirfenidone treatment groups than in those taking placebo. Dosage reduction or interruption for gastrointestinal events was required in 18.5% of patients in the 2403 mg/day group, as compared to 5.8% of patients in the placebo group; 2.2% of patients in the pirfenidone 2403 mg/day group discontinued treatment due to a gastrointestinal event, as compared to 1.0% in the placebo group. The most common (>2%) gastrointestinal events that led to dosage reduction or interruption were nausea, diarrhea, vomiting, and dyspepsia. The incidence of gastrointestinal events was highest early in the course of treatment (with highest incidence occurring during the initial 3 months) and decreased over time. Dosage modifications may be necessary in some cases of gastrointestinal adverse reactions [see Dosage and Administration (2.3)].
•
**Elevated liver enzymes and drug-induced liver injury:** ALT, AST, and bilirubin elevations have occurred with pirfenidone including cases of drug-induced liver injury. In the postmarketing setting, non-serious and serious cases of drug-induced liver injury, including severe liver injury with fatal outcomes, have been reported. Monitor ALT, AST, and bilirubin before and during treatment. Temporary dosage reductions or discontinuations may be required. (2.1, 5.1)
•
**Photosensitivity and rash:** Photosensitivity and rash have been noted with pirfenidone. Avoid exposure to sunlight and sunlamps. Wear sunscreen and protective clothing daily. Temporary dosage reductions or discontinuations may be required. (5.2)
•
**Severe Cutaneous Adverse Reactions (SCAR):** Stevens-Johnson syndrome (SJS), toxic epidermal necrolysis (TEN), and drug reactions with eosinophilia and systemic symptoms (DRESS) have been reported in association with the use of pirfenidone in the postmarketing setting. Interrupt pirfenidone in case of signs or symptoms of SCAR. Permanently discontinue pirfenidone if a SCAR is confirmed. (5.3)
•
**Gastrointestinal disorders:** Nausea, vomiting, diarrhea, dyspepsia, gastro-esophageal reflux disease, and abdominal pain have occurred with pirfenidone. Temporary dosage reductions or discontinuations may be required. (5.4)
NONCLINICAL TOXICOLOGY SECTION
13 NONCLINICAL TOXICOLOGY
13.1 Carcinogenesis, Mutagenesis, Impairment of Fertility
Carcinogenesis
Long-term studies were conducted in mice and rats with admixture of pirfenidone to the diet to evaluate its carcinogenic potential.
In a 24-month carcinogenicity study in B6C3F1 mice, pirfenidone caused statistically significant dose-related increases of the combination of hepatocellular adenoma and carcinoma and hepatoblastoma in male mice at doses of 800 mg/kg and above (AUC exposure approximately 0.4 times adult exposure at the MRDD). There were statistically significant dose-related increases of the combination of hepatocellular adenoma and carcinoma in female mice at doses of 2000 mg/kg and above (AUC exposure approximately 0.7 times adult exposure at the MRDD).
In a 24-month carcinogenicity study in Fischer rats, pirfenidone caused statistically significant dose-related increases of the combination of hepatocellular adenoma and carcinoma in male rats at doses of 750 mg/kg and above (AUC exposure approximately 1.9 times adult exposure at the MRDD). There were statistically significant increases of the combination of hepatocellular adenoma and carcinoma and the combination of uterine adenocarcinoma and adenoma at a dose of 1500 mg/kg/day (AUC exposure approximately 3.0 times adult exposure at the MRDD).
The relevance of these tumor findings in rodents to humans is unknown.
Mutagenesis
Pirfenidone was not mutagenic or clastogenic in the following tests: mutagenicity tests in bacteria, a chromosomal aberration test in Chinese hamster lung cells, and a micronucleus test in mice.
Impairment of Fertility
Pirfenidone had no effects on fertility and reproductive performance in rats at dosages up to 1000 mg/kg/day (approximately 3 times the MRDD in adults on a mg/m2 basis).
CLINICAL STUDIES SECTION
14 CLINICAL STUDIES
The efficacy of pirfenidone was evaluated in patients with IPF in three phase 3, randomized, double-blind, placebo-controlled, multicenter trials (Studies 1, 2, and 3).
Study 1 was a 52-week trial comparing pirfenidone 2403 mg/day (n=278) versus placebo (n=277) in patients with IPF. Study 2 and Study 3 were nearly identical to each other in design, with few exceptions, including an intermediate dose treatment arm in Study 2. Study 2 compared treatment with either pirfenidone 2403 mg/day (n=174) or pirfenidone 1197 mg/day (n=87) to placebo (n=174), while Study 3 compared pirfenidone 2403 mg/day (n=171) to placebo (n=173). Study drug was administered three times daily with food for a minimum of 72 weeks. Patients continued on treatment until the last patient completed 72 weeks of treatment, which included observations to approximately 120 weeks of study treatment. The primary endpoint was the change in percent predicted forced vital capacity (%FVC) from baseline to study end, measured at 52 weeks in Study 1, and at 72 weeks in Studies 2 and 3.
Studies 1, 2 and 3 enrolled adult patients who had a clinical and radiographic diagnosis of IPF (with or without accompanying surgical lung biopsy), without evidence or suspicion of an alternative diagnosis for interstitial lung disease. Eligible patients were to have %FVC greater than or equal to 50% at baseline and a percent predicted diffusing capacity of the lungs for carbon monoxide (%DLCO) greater than or equal to 30% (Study 1) or 35% (Studies 2 and 3) at baseline. In all three trials, over 80% of patients completed study treatment.
A total of 1247 patients with IPF were randomized to receive pirfenidone 2403 mg/day (n=623) or placebo (n=624) in these three trials. Baseline characteristics were generally balanced across treatment groups. The study population ranged from 40 to 80 years of age (mean age 67 years). Most patients were male (74%), white (95%), and current or former smokers (65%). Approximately 93% of patients met criteria for definite IPF on high resolution computed tomography (HRCT). Baseline mean %FVC and %DLCO were 72% and 46%, respectively. Approximately 15% subjects discontinued from each treatment group.
Change from Baseline in Percent Predicted Forced Vital Capacity
In Study 1, the primary efficacy analysis for the change in %FVC from baseline to Week 52 demonstrated a statistically significant treatment effect of pirfenidone 2403 mg/day (n=278) compared with placebo (n=277) using a rank ANCOVA with the lowest rank imputation for missing data due to death. In Study 2, there was a statistically significant difference at Week 72 for the change in %FVC from baseline. In Study 3, there was no statistically significant difference at Week 72 for the change in %FVC from baseline.
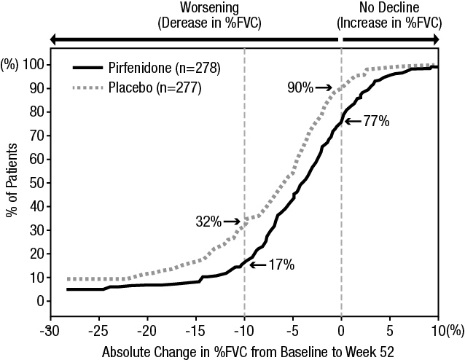
Figure 1presents the cumulative distribution for all cut-offs for the change from baseline in %FVC at Week 52 for Study 1. For all categorical declines in lung function, the proportion of patients declining was lower on pirfenidone than on placebo. Study 2 showed similar results.
Figure 1. Cumulative Distribution of Patients by Change in Percent Predicted FVC from Baseline to Week 52 (Study 1). The Dashed Lines Indicate ≥10% Decline or ≥0% Decline.
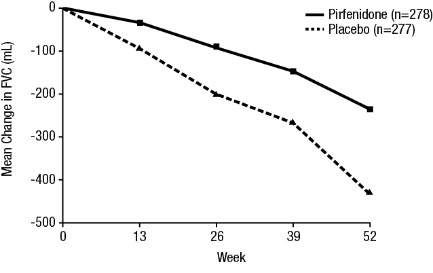
Mean Change from Baseline in FVC (mL)
In Study 1, a reduction in the mean decline in FVC (in mL) was observed in patients receiving pirfenidone 2403 mg/day (-235 mL) compared to placebo (-428 mL) (mean treatment difference 193 mL) at Week 52 (seeFigure 2). In Study 2, a reduction in the decline in FVC volume was also observed in patients receiving pirfenidone 2403 mg/day compared with placebo (mean treatment difference 157 mL) at Week 72. There was no statistically significant difference in decline in FVC volume seen in Study 3.
**Figure 2. Mean Change from Baseline in Forced Vital Capacity (Study 1)**
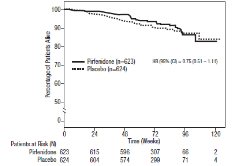
Survival
Survival was evaluated for pirfenidone compared to placebo in Studies 1, 2, and 3 as an exploratory analysis to support the primary endpoint (FVC). All- cause mortality was assessed over the study duration and available follow-up period, irrespective of cause of death and whether patients continued treatment. All-cause mortality did not show a statistically significant difference (seeFigure 3).
**Figure 3. Kaplan-Meier Estimates of All-Cause Mortality at Vital Status – End of Study: Studies 1, 2, and 3**
HOW SUPPLIED SECTION
16 HOW SUPPLIED/STORAGE AND HANDLING
Pirfenidone tablets are available as follows:
267 mg (yellow): oval, biconvex, film-coated tablet, debossed with SD267 on one side.
NDC 0781-8085-32, bottle of 270 tablets total, with a child-resistant closure
801 mg (dark pink): oval, biconvex, film-coated tablet, debossed with SD801 on one side.
NDC 0781-8086-92, bottle of 90 tablets, with a child-resistant closure
Store at 25°C (77°F); excursions permitted to 15°C to 30°C (59°F to 86°F) [see USP Controlled Room Temperature].
Keep the bottle tightly closed. Do not use if the seal over the bottle opening is broken or missing. Safely throw away any pirfenidone tablets that is out of date or no longer needed.
INFORMATION FOR PATIENTS SECTION
17 PATIENT COUNSELING INFORMATION
Advise the patient to read the FDA-approved patient labeling (Patient Information).
Liver Enzyme Elevations
Advise patients that they may be required to undergo liver function testing periodically. Instruct patients to immediately report any symptoms of a liver problem (e.g., skin or the white of eyes turn yellow, urine turns dark or brown [tea colored], pain on the right side of stomach, bleed or bruise more easily than normal, lethargy) [see Warnings and Precautions (5.1)].
Photosensitivity Reaction or Rash
Advise patients to avoid or minimize exposure to sunlight (including sunlamps) during use of pirfenidone tablets because of concern for photosensitivity reactions or rash. Instruct patients to use a sunblock and to wear clothing that protects against sun exposure. Instruct patients to report symptoms of photosensitivity reaction or rash to their physician. Temporary dosage reductions or discontinuations may be required [see Warnings and Precautions (5.2)].
Severe Cutaneous Adverse Reactions
Advise patients about signs and symptoms of severe cutaneous adverse reactions (SCAR). Advise patients to contact their healthcare provider immediately if they experience signs and symptoms of SCAR [see Warnings and Precautions (5.3)].
Gastrointestinal Events
Instruct patients to report symptoms of persistent gastrointestinal effects including nausea, diarrhea, dyspepsia, vomiting, gastro-esophageal reflux disease, and abdominal pain. Temporary dosage reductions or discontinuations may be required [see Warnings and Precautions (5.4)].
Smokers
Encourage patients to stop smoking prior to treatment with pirfenidone and to avoid smoking when using pirfenidone tablets [see Clinical Pharmacology (12.3)].
Take with Food
Instruct patients to take pirfenidone tablets with food to help decrease nausea and dizziness.