
Abacavir
These highlights do not include all the information needed to use ABACAVIR TABLETS safely and effectively. See full prescribing information for ABACAVIR TABLETS. ABACAVIR tablets, for oral use Initial U.S. Approval: 1998
5409c8ed-17c3-454f-bb88-d597ba8f84ec
HUMAN PRESCRIPTION DRUG LABEL
Apr 15, 2023
Aurobindo Pharma Limited
DUNS: 650082092
Products 1
Detailed information about drug products covered under this FDA approval, including NDC codes, dosage forms, ingredients, and administration routes.
Abacavir
Product Details
FDA regulatory identification and product classification information
FDA Identifiers
Product Classification
Product Specifications
INGREDIENTS (10)
Drug Labeling Information
Warnings And Precautions Section
5 WARNINGS AND PRECAUTIONS
5.1 Hypersensitivity Reactions
Serious and sometimes fatal hypersensitivity reactions have occurred with abacavir. These hypersensitivity reactions have included multi-organ failure and anaphylaxis and typically occurred within the first 6 weeks of treatment with abacavir (median time to onset was 9 days); although abacavir hypersensitivity reactions have occurred any time during treatment [see Adverse Reactions (6.1)]. Patients who carry the HLA-B5701 allele are at a higher risk of abacavir hypersensitivity reactions; although, patients who do not carry the HLA-B5701 allele have developed hypersensitivity reactions. Hypersensitivity to abacavir was reported in approximately 206 (8%) of 2,670 patients in 9 clinical trials with abacavir-containing products where HLA-B5701 screening was not performed. The incidence of suspected abacavir hypersensitivity reactions in clinical trials was 1% when subjects carrying the HLA-B5701 allele were excluded. In any patient treated with abacavir, the clinical diagnosis of hypersensitivity reaction must remain the basis of clinical decision making.
Due to the potential for severe, serious, and possibly fatal hypersensitivity reactions with abacavir:
- All patients should be screened for the HLA-B5701 allele prior to initiating therapy with abacavir or reinitiation of therapy with abacavir, unless patients have a previously documented HLA-B5701 allele assessment.
- Abacavir is contraindicated in patients with a prior hypersensitivity reaction to abacavir and in HLA-B*5701-positive patients.
- Before starting abacavir, review medical history for prior exposure to any abacavir-containing product. NEVER restart abacavir or any other abacavir-containing product following a hypersensitivity reaction to abacavir, regardless of HLA-B*5701 status.
- To reduce the risk of a life-threatening hypersensitivity reaction, regardless of HLA-B*5701 status, discontinue abacavir immediately if a hypersensitivity reaction is suspected, even when other diagnoses are possible (e.g., acute onset respiratory diseases such as pneumonia, bronchitis, pharyngitis, or influenza; gastroenteritis; or reactions to other medications).
- If a hypersensitivity reaction cannot be ruled out, do not restart abacavir or any other abacavir-containing products because more severe symptoms which may include life-threatening hypotension and death, can occur within hours.
- If a hypersensitivity reaction is ruled out, patients may restart abacavir. Rarely, patients who have stopped abacavir for reasons other than symptoms of hypersensitivity have also experienced life-threatening reactions within hours of reinitiating abacavir therapy. Therefore, reintroduction of abacavir or any other abacavir-containing product is recommended only if medical care can be readily accessed.
- A Medication Guide and Warning Card that provide information about recognition of hypersensitivity reactions should be dispensed with each new prescription and refill.
5.2 Lactic Acidosis and Severe Hepatomegaly with Steatosis
Lactic acidosis and severe hepatomegaly with steatosis, including fatal cases, have been reported with the use of nucleoside analogues, including abacavir. A majority of these cases have been in women. Female sex and obesity may be risk factors for the development of lactic acidosis and severe hepatomegaly with steatosis in patients treated with antiretroviral nucleoside analogues. Treatment with abacavir should be suspended in any patient who develops clinical or laboratory findings suggestive of lactic acidosis or pronounced hepatotoxicity, which may include hepatomegaly and steatosis even in the absence of marked transaminase elevations.
5.3 Immune Reconstitution Syndrome
Immune reconstitution syndrome has been reported in patients treated with combination antiretroviral therapy, including abacavir. During the initial phase of combination antiretroviral treatment, patients whose immune systems respond may develop an inflammatory response to indolent or residual opportunistic infections (such as Mycobacterium avium infection, cytomegalovirus, Pneumocystis jirovecii pneumonia [PCP], or tuberculosis), which may necessitate further evaluation and treatment.
Autoimmune disorders (such as Graves’ disease, polymyositis, and Guillain- Barré syndrome) have also been reported to occur in the setting of immune reconstitution; however, the time to onset is more variable and can occur many months after initiation of treatment.
5.4 Myocardial Infarction
Several prospective, observational, epidemiological studies have reported an association with the use of abacavir and the risk of myocardial infarction (MI). Meta-analyses of randomized, controlled, clinical trials have observed no excess risk of MI in abacavir-treated subjects as compared with control subjects. To date, there is no established biological mechanism to explain a potential increase in risk. In totality, the available data from the observational studies and from controlled clinical trials show inconsistency; therefore, evidence for a causal relationship between abacavir treatment and the risk of MI is inconclusive.
As a precaution, the underlying risk of coronary heart disease should be considered when prescribing antiretroviral therapies, including abacavir, and action taken to minimize all modifiable risk factors (e.g., hypertension, hyperlipidemia, diabetes mellitus, smoking).
- Lactic acidosis and severe hepatomegaly with steatosis, including fatal cases, have been reported with the use of nucleoside analogues. (5.2)
- Immune reconstitution syndrome has been reported in patients treated with combination antiretroviral therapy. (5.3)
Adverse Reactions Section
6 ADVERSE REACTIONS
The following adverse reactions are discussed in other sections of the labeling:
- Serious and sometimes fatal hypersensitivity reactions [see Boxed Warning, Warnings and Precautions (5.1)].
- Lactic acidosis and severe hepatomegaly with steatosis [see Warnings and Precautions (5.2)].
- Immune reconstitution syndrome [see Warnings and Precautions (5.3)].
- Myocardial infarction [see Warnings and Precautions (5.4)].
6.1 Clinical Trials Experience in Adult Subjects
Because clinical trials are conducted under widely varying conditions, adverse reaction rates observed in the clinical trials of a drug cannot be directly compared with rates in the clinical trials of another drug and may not reflect the rates observed in clinical practice.
Serious and Fatal Abacavir-Associated Hypersensitivity Reactions
In clinical trials, serious and sometimes fatal hypersensitivity reactions have occurred with abacavir [see Boxed Warning, Warnings and Precautions (5.1)]. These reactions have been characterized by 2 or more of the following signs or symptoms: (1) fever; (2) rash; (3) gastrointestinal symptoms (including nausea, vomiting, diarrhea, or abdominal pain); (4) constitutional symptoms (including generalized malaise, fatigue, or achiness); (5) respiratory symptoms (including dyspnea, cough, or pharyngitis). Almost all abacavir hypersensitivity reactions include fever and/or rash as part of the syndrome.
Other signs and symptoms have included lethargy, headache, myalgia, edema, arthralgia, and paresthesia. Anaphylaxis, liver failure, renal failure, hypotension, adult respiratory distress syndrome, respiratory failure, myolysis, and death have occurred in association with these hypersensitivity reactions. Physical findings have included lymphadenopathy, mucous membrane lesions (conjunctivitis and mouth ulcerations), and maculopapular or urticarial rash (although some patients had other types of rashes and others did not have a rash). There were reports of erythema multiforme. Laboratory abnormalities included elevated liver chemistries, elevated creatine phosphokinase, elevated creatinine, and lymphopenia, and abnormal chest x-ray findings (predominantly infiltrates, which were localized).
Additional Adverse Reactions with Use of Abacavir
Therapy-Naive Adults: Treatment-emergent clinical adverse reactions (rated by the investigator as moderate or severe) with a greater than or equal to 5% frequency during therapy with abacavir 300 mg twice daily, lamivudine 150 mg twice daily, and efavirenz 600 mg daily compared with zidovudine 300 mg twice daily, lamivudine 150 mg twice daily, and efavirenz 600 mg daily from CNA30024 are listed in Table 2.
Table 2. Treatment-Emergent (All Causality) Adverse Reactions of at Least Moderate Intensity (Grades 2 to 4, Greater than or Equal to 5% Frequency) in Therapy-Naive Adults (CNA30024a) through 48 Weeks of Treatment|
a This trial used double-blind ascertainment of suspected hypersensitivity
reactions. During the blinded portion of the trial, suspected hypersensitivity
to abacavir was reported by investigators in 9% of 324 subjects in the
abacavir group and 3% of 325 subjects in the zidovudine group. | ||
|
Adverse Reaction |
Abacavir plus |
Zidovudine plus |
|
Dreams/sleep disorders |
10% |
10% |
|
Drug hypersensitivity |
9% |
<1%b |
|
Headaches/migraine |
7% |
11% |
|
Nausea |
7% |
11% |
|
Fatigue/malaise |
7% |
10% |
|
Diarrhea |
7% |
6% |
|
Rashes |
6% |
12% |
|
Abdominal pain/gastritis/gastrointestinal signs and symptoms |
6% |
8% |
|
Depressive disorders |
6% |
6% |
|
Dizziness |
6% |
6% |
|
Musculoskeletal pain |
6% |
5% |
|
Bronchitis |
4% |
5% |
|
Vomiting |
2% |
9% |
Treatment-emergent clinical adverse reactions (rated by the investigator as moderate or severe) with a greater than or equal to 5% frequency during therapy with abacavir 300 mg twice daily, lamivudine 150 mg twice daily, and zidovudine 300 mg twice daily compared with indinavir 800 mg 3 times daily, lamivudine 150 mg twice daily, and zidovudine 300 mg twice daily from CNA3005 are listed in Table 3.
Table 3. Treatment-Emergent (All Causality) Adverse Reactions of at Least Moderate Intensity (Grades 2 to 4, Greater than or Equal to 5% Frequency) in Therapy-Naive Adults (CNA3005) through 48 Weeks of Treatment|
Adverse Reaction |
Abacavir plus |
Indinavir plus |
|
Nausea |
19% |
17% |
|
Headache |
13% |
9% |
|
Malaise and fatigue |
12% |
12% |
|
Nausea and vomiting |
10% |
10% |
|
Hypersensitivity reaction |
8% |
2% |
|
Diarrhea |
7% |
5% |
|
Fever and/or chills |
6% |
3% |
|
Depressive disorders |
6% |
4% |
|
Musculoskeletal pain |
5% |
7% |
|
Skin rashes |
5% |
4% |
|
Ear/nose/throat infections |
5% |
4% |
|
Viral respiratory infections |
5% |
5% |
|
Anxiety |
5% |
3% |
|
Renal signs/symptoms |
<1% |
5% |
|
Pain (non-site-specific) |
<1% |
5% |
Five subjects receiving abacavir in CNA3005 experienced worsening of pre- existing depression compared with none in the indinavir arm. The background rates of pre-existing depression were similar in the 2 treatment arms.
Abacavir Once Daily versus Abacavir Twice Daily (CNA30021): Treatment-emergent clinical adverse reactions (rated by the investigator as at least moderate) with a greater than or equal to 5% frequency during therapy with abacavir 600 mg once daily or abacavir 300 mg twice daily, both in combination with lamivudine 300 mg once daily and efavirenz 600 mg once daily from CNA30021, were similar. For hypersensitivity reactions, subjects receiving abacavir once daily showed a rate of 9% in comparison with a rate of 7% for subjects receiving abacavir twice daily. However, subjects receiving abacavir 600 mg once daily experienced a significantly higher incidence of severe drug hypersensitivity reactions and severe diarrhea compared with subjects who received abacavir 300 mg twice daily. Five percent (5%) of subjects receiving abacavir 600 mg once daily had severe drug hypersensitivity reactions compared with 2% of subjects receiving abacavir 300 mg twice daily. Two percent (2%) of subjects receiving abacavir 600 mg once daily had severe diarrhea while none of the subjects receiving abacavir 300 mg twice daily had this event.
Laboratory Abnormalities: Laboratory abnormalities (Grades 3 to 4) in therapy- naive adults during therapy with abacavir 300 mg twice daily, lamivudine 150 mg twice daily, and efavirenz 600 mg daily compared with zidovudine 300 mg twice daily, lamivudine 150 mg twice daily, and efavirenz 600 mg daily from CNA30024 are listed in Table 4.
Table 4. Laboratory Abnormalities (Grades 3 to 4) in Therapy-Naive Adults (CNA30024) through 48 Weeks of Treatment|
ULN = Upper limit of normal. | ||
|
Grade 3/4 |
Abacavir plus |
Zidovudine plus |
|
Elevated CPK (>4 X ULN) |
8% |
8% |
|
Elevated ALT (>5 X ULN) |
6% |
6% |
|
Elevated AST (>5 X ULN) |
6% |
5% |
|
Hypertriglyceridemia (>750 mg/dL) |
6% |
5% |
|
Hyperamylasemia (>2 X ULN) |
4% |
5% |
|
Neutropenia (ANC <750/mm3) |
2% |
4% |
|
Anemia (Hgb ≤6.9 gm/dL) |
<1% |
2% |
|
Thrombocytopenia (Platelets <50,000/mm3) |
1% |
<1% |
|
Leukopenia (WBC ≤1,500/mm3) |
<1% |
2% |
Laboratory abnormalities in CNA3005 are listed in Table 5.
Table 5. Treatment-Emergent Laboratory Abnormalities (Grades 3 to 4) in CNA3005|
ULN = Upper limit of normal. | ||
|
Grade 3/4 Laboratory Abnormalities | ||
|
Abacavir plus |
Indinavir plus | |
|
Elevated CPK (>4 x ULN) |
18 (7%) |
18 (7%) |
|
ALT (>5 x ULN) |
16 (6%) |
16 (6%) |
|
Neutropenia (<750/mm3) |
13 (5%) |
13 (5%) |
|
Hypertriglyceridemia (>750 mg/dL) |
5 (2%) |
3 (1%) |
|
Hyperamylasemia (>2 x ULN) |
5 (2%) |
1 (<1%) |
|
Hyperglycemia (>13.9 mmol/L) |
2 (<1%) |
2 (<1%) |
|
Anemia (Hgb ≤6.9 g/dL) |
0 (0%) |
3 (1%) |
The frequencies of treatment-emergent laboratory abnormalities were comparable between treatment groups in CNA30021.
6.2 Clinical Trials Experience in Pediatric Subjects
Therapy-Experienced Pediatric Subjects (Twice-Daily Dosing)
Treatment-emergent clinical adverse reactions (rated by the investigator as moderate or severe) with a greater than or equal to 5% frequency during therapy with abacavir 8 mg per kg twice daily, lamivudine 4 mg per kg twice daily, and zidovudine 180 mg per m2 twice daily compared with lamivudine 4 mg per kg twice daily and zidovudine 180 mg per m2 twice daily from CNA3006 are listed in Table 6.
Table 6. Treatment-Emergent (All Causality) Adverse Reactions of at Least Moderate Intensity (Grades 2 to 4, Greater than or Equal to 5% Frequency) in Therapy-Experienced Pediatric Subjects (CNA3006) through 16 Weeks of Treatment|
Adverse Reaction |
Abacavir plus Lamivudine plus Zidovudine |
Lamivudine plus Zidovudine |
|
Fever and/or chills |
9% |
7% |
|
Nausea and vomiting |
9% |
2% |
|
Skin rashes |
7% |
1% |
|
Ear/nose/throat infections |
5% |
1% |
|
Pneumonia |
4% |
5% |
|
Headache |
1% |
5% |
Laboratory Abnormalities: In CNA3006, laboratory abnormalities (anemia, neutropenia, liver function test abnormalities, and CPK elevations) were observed with similar frequencies as in a trial of therapy-naive adults (CNA30024). Mild elevations of blood glucose were more frequent in pediatric subjects receiving abacavir (CNA3006) as compared with adult subjects (CNA30024).
Other Adverse Events
In addition to adverse reactions and laboratory abnormalities reported in Tables 2, 3, 4, 5, and 6, other adverse reactions observed in the expanded access program were pancreatitis and increased GGT.
Pediatric Subjects Once-Daily versus Twice-Daily Dosing (COL105677): The safety of once-daily compared with twice-daily dosing of abacavir was assessed in the ARROW trial. Primary safety assessment in the ARROW trial was based on Grade 3 and Grade 4 adverse events. The frequency of Grade 3 and 4 adverse events was similar among subjects randomized to once-daily dosing compared with subjects randomized to twice-daily dosing. One event of Grade 4 hepatitis in the once-daily cohort was considered as uncertain causality by the investigator and all other Grade 3 or 4 adverse events were considered not related by the investigator.
6.3 Postmarketing Experience
The following adverse reactions have been identified during postmarketing use of abacavir. Because these reactions are reported voluntarily from a population of unknown size, it is not always possible to reliably estimate their frequency or establish a causal relationship to drug exposures.
Body as a Whole
Redistribution/accumulation of body fat.
Cardiovascular
Myocardial infarction.
Hepatic
Lactic acidosis and hepatic steatosis [see Warnings and Precautions (5.2)].
Skin
Suspected Stevens-Johnson syndrome (SJS) and toxic epidermal necrolysis (TEN) have been reported in patients receiving abacavir primarily in combination with medications known to be associated with SJS and TEN, respectively. Because of the overlap of clinical signs and symptoms between hypersensitivity to abacavir and SJS and TEN, and the possibility of multiple drug sensitivities in some patients, abacavir should be discontinued and not restarted in such cases.
There have also been reports of erythema multiforme with abacavir use [see Adverse Reactions (6.1)].
- The most commonly reported adverse reactions of at least moderate intensity (incidence greater than or equal to 10%) in adult HIV-1 clinical trials were nausea, headache, malaise and fatigue, nausea and vomiting, and dreams/sleep disorders. (6.1)
- The most commonly reported adverse reactions of at least moderate intensity (incidence greater than or equal to 5%) in pediatric HIV-1 clinical trials were fever and/or chills, nausea and vomiting, skin rashes, and ear/nose/throat infections. (6.2)
To report SUSPECTED ADVERSE REACTIONS, contact Aurobindo Pharma USA, Inc. at 1-866-850-2876 or FDA at 1-800-FDA-1088 orwww.fda.gov/medwatch.
Drug Interactions Section
7 DRUG INTERACTIONS
7.1 Methadone
In a trial of 11 HIV-1-infected subjects receiving methadone-maintenance therapy with 600 mg of abacavir twice daily (twice the currently recommended dose), oral methadone clearance increased [see Clinical Pharmacology (12.3)]. This alteration will not result in a methadone dose modification in the majority of patients; however, an increased methadone dose may be required in a small number of patients.
7.2 Riociguat
Coadministration with fixed-dose abacavir/dolutegravir/lamivudine resulted in increased riociguat exposure, which may increase the risk of riociguat adverse reactions [see Clinical Pharmacology (12.3)]. The riociguat dose may need to be reduced. See full prescribing information for ADEMPAS (riociguat).
- Methadone: An increased methadone dose may be required in a small number of patients. (7.1)
- Riociguat: The riociguat dose may need to be reduced. (7.2)
Dosage & Administration Section
2 DOSAGE AND ADMINISTRATION
2.1 Screening for HLA-B*5701 Allele prior to Starting Abacavir Tablets
Screen for the HLA-B*5701 allele prior to initiating therapy with abacavir tablets [see Boxed Warning, Warnings and Precautions (5.1)].
2.2 Recommended Dosage for Adult Patients
The recommended dosage of abacavir tablets for adults is 600 mg daily, administered orally as either 300 mg twice daily or 600 mg once daily, in combination with other antiretroviral agents.
2.3 Recommended Dosage for Pediatric Patients
Abacavir tablets are available as scored tablet for HIV-1-infected pediatric patients weighing greater than or equal to 14 kg for whom a solid dosage form is appropriate. Before prescribing abacavir tablets, children should be assessed for the ability to swallow tablets. If a child is unable to reliably swallow abacavir tablets, the oral solution formulation should be prescribed. The recommended oral dosage of abacavir tablets for HIV-1-infected pediatric patients is presented in Table 1.
Table 1. Dosing Recommendations for Abacavir Scored Tablets in Pediatric Patients|
Weight |
Once-Daily |
Twice-Daily Dosing Regimen | ||
|
AM Dose |
PM Dose |
Total | ||
|
14 to <20 |
1 tablet |
½ tablet |
½ tablet |
300 mg |
|
1½ tablets |
½ tablet |
1 tablet |
450 mg |
|
≥25 |
2 tablets |
1 tablet |
1 tablet |
600 mg |
a Data regarding the efficacy of once-daily dosing is limited to subjects who transitioned from twice-daily dosing to once-daily dosing after 36 weeks of treatment [see Clinical Studies (14.2)].
2.4 Recommended Dosage for Patients with Hepatic Impairment
The recommended dose of abacavir tablets in patients with mild hepatic impairment (Child-Pugh Class A) is 200 mg twice daily. To enable dose reduction, abacavir oral solution (10 mL twice daily) should be used for the treatment of these patients. The safety, efficacy, and pharmacokinetic properties of abacavir have not been established in patients with moderate to severe hepatic impairment; therefore, abacavir tablets are contraindicated in these patients.
- Before initiating abacavir, screen for the HLA-B*5701 allele. (2.1)
- Adults: 600 mg daily, administered as either 300 mg twice daily or 600 mg once daily. (2.2)
- Pediatric Patients Aged 3 Months and Older: Administered either once or twice daily. Dose should be calculated on body weight (kg) and should not exceed 600 mg daily. (2.3)
- Patients with Hepatic Impairment: Mild hepatic impairment – 200 mg twice daily. (2.4)
Description Section
11 DESCRIPTION
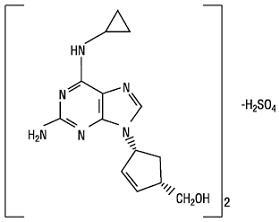
Abacavir sulfate is a synthetic carbocyclic nucleoside analogue with inhibitory activity against HIV-1. The chemical name of abacavir sulfate is (1S,cis)-4-[2-amino-6-(cyclopropylamino)-9H-purin-9-yl]-2-cyclopentene-1-methanol sulfate (salt) (2:1). Abacavir sulfate is the enantiomer with 1S, 4R absolute configuration on the cyclopentene ring. It has a molecular formula of (C14H18N6O)2•H2SO4 and a molecular weight of 670.76 g per mol. It has the following structural formula:
Abacavir sulfate USP is a white to off-white solid and is soluble in water.
Abacavir tablets USP are for oral administration. Each tablet contains abacavir sulfate USP equivalent to 300 mg of abacavir as active ingredient and the following inactive ingredients: colloidal silicon dioxide, magnesium stearate, microcrystalline cellulose, and sodium starch glycolate. The tablets are coated with a film that is made of hypromellose, iron oxide yellow, polysorbate 80, titanium dioxide, and triacetin.
In vivo, abacavir sulfate dissociates to its free base, abacavir. Dosages are expressed in terms of abacavir.
Nonclinical Toxicology Section
13 NONCLINICAL TOXICOLOGY
13.1 Carcinogenesis, Mutagenesis, Impairment of Fertility
Carcinogenicity
Abacavir was administered orally at 3 dosage levels to separate groups of mice and rats in 2-year carcinogenicity studies. Results showed an increase in the incidence of malignant and non-malignant tumors. Malignant tumors occurred in the preputial gland of males and the clitoral gland of females of both species, and in the liver of female rats. In addition, non-malignant tumors also occurred in the liver and thyroid gland of female rats. These observations were made at systemic exposures in the range of 6 to 32 times the human exposure at the recommended dose of 600 mg.
Mutagenicity
Abacavir induced chromosomal aberrations both in the presence and absence of metabolic activation in an in vitro cytogenetic study in human lymphocytes. Abacavir was mutagenic in the absence of metabolic activation, although it was not mutagenic in the presence of metabolic activation in an L5178Y mouse lymphoma assay. Abacavir was clastogenic in males and not clastogenic in females in an in vivo mouse bone marrow micronucleus assay.
Abacavir was not mutagenic in bacterial mutagenicity assays in the presence and absence of metabolic activation.
Impairment of Fertility
Abacavir did not affect male or female fertility in rats at a dose associated with exposures (AUC) approximately 3.3 times (male) or 4.1 times (female) those in humans at the clinically recommended dose.
13.2 Animal Toxicology and/or Pharmacology
Myocardial degeneration was found in mice and rats following administration of abacavir for 2 years. The systemic exposures were equivalent to 7 to 24 times the expected systemic exposure in humans at a dose of 600 mg. The clinical relevance of this finding has not been determined.
Clinical Studies Section
14 CLINICAL STUDIES
14.1 Adult Trials
Therapy-Naive Adults
CNA30024 was a multicenter, double-blind, controlled trial in which 649 HIV-1-infected, therapy-naive adults were randomized and received either abacavir (300 mg twice daily), lamivudine (150 mg twice daily), and efavirenz (600 mg once daily); or zidovudine (300 mg twice daily), lamivudine (150 mg twice daily), and efavirenz (600 mg once daily). The duration of double-blind treatment was at least 48 weeks. Trial participants were male (81%), white (51%), black (21%), and Hispanic (26%). The median age was 35 years; the median pretreatment CD4+ cell count was 264 cells per mm3, and median plasma HIV-1 RNA was 4.79 log10 copies per mL. The outcomes of randomized treatment are provided in Table 7.
Table 7. Outcomes of Randomized Treatment through Week 48 (CNA30024)|
a Subjects achieved and maintained confirmed HIV-1 RNA less than or equal to
50 copies per mL (less than 400 copies per mL) through Week 48 (Roche AMPLICOR
Ultrasensitive HIV-1 MONITOR standard test 1 PCR). | ||
|
Outcome |
Abacavir plus |
Zidovudine plus |
|
Respondera |
69% (73%) |
69% (71%) |
|
Virologic failuresb |
6% |
4% |
|
Discontinued due to adverse reactions |
14% |
16% |
|
Discontinued due to other reasonsc |
10% |
11% |
After 48 weeks of therapy, the median CD4+ cell count increases from baseline were 209 cells per mm3 in the group receiving abacavir and 155 cells per mm3 in the zidovudine group. Through Week 48, 8 subjects (2%) in the group receiving abacavir (5 CDC classification C events and 3 deaths) and 5 subjects (2%) on the zidovudine arm (3 CDC classification C events and 2 deaths) experienced clinical disease progression.
CNA3005 was a multicenter, double-blind, controlled trial in which 562 HIV-1-infected, therapy-naive adults were randomized to receive either abacavir (300 mg twice daily) plus COMBIVIR (lamivudine 150 mg/zidovudine 300 mg twice daily), or indinavir (800 mg 3 times a day) plus COMBIVIR twice daily. The trial was stratified at randomization by pre-entry plasma HIV-1 RNA 10,000 to 100,000 copies per mL and plasma HIV-1 RNA greater than 100,000 copies per mL. Trial participants were male (87%), white (73%), black (15%), and Hispanic (9%). At baseline the median age was 36 years; the median baseline CD4+ cell count was 360 cells per mm3, and median baseline plasma HIV-1 RNA was 4.8 log10 copies per mL. Proportions of subjects with plasma HIV-1 RNA less than 400 copies per mL (using Roche AMPLICOR HIV-1 MONITOR Test) through 48 weeks of treatment are summarized in Table 8.
Table 8. Outcomes of Randomized Treatment through Week 48 (CNA3005)|
a Subjects achieved and maintained confirmed HIV-1 RNA less than 400 copies
per mL. | ||
|
Outcome |
Abacavir plus |
Indinavir plus |
|
Respondera |
49% |
50% |
|
Virologic failureb |
31% |
28% |
|
Discontinued due to adverse reactions |
10% |
12% |
|
Discontinued due to other reasonsc |
11% |
10% |
Treatment response by plasma HIV-1 RNA strata is shown in Table 9.
Table 9. Proportions of Responders through Week 48 by Screening Plasma HIV-1 RNA Levels (CNA3005)|
Screening |
Abacavir plus |
Indinavir plus | ||
|
<400 copies/mL |
n |
<400 copies/mL |
n | |
|
≥10,000 to ≤100,000
|
50% |
166 |
48% |
165 |
In subjects with baseline viral load greater than 100,000 copies per mL, percentages of subjects with HIV-1 RNA levels less than 50 copies per mL were 31% in the group receiving abacavir versus 45% in the group receiving indinavir.
Through Week 48, an overall mean increase in CD4+ cell count of about 150 cells per mm3 was observed in both treatment arms. Through Week 48, 9 subjects (3.4%) in the group receiving abacavir (6 CDC classification C events and 3 deaths) and 3 subjects (1.5%) in the group receiving indinavir (2 CDC classification C events and 1 death) experienced clinical disease progression.
CNA30021 was an international, multicenter, double-blind, controlled trial in which 770 HIV-1-infected, therapy-naive adults were randomized and received either abacavir 600 mg once daily or abacavir 300 mg twice daily, both in combination with lamivudine 300 mg once daily and efavirenz 600 mg once daily. The double-blind treatment duration was at least 48 weeks. Trial participants had a mean age of 37 years; were male (81%), white (54%), black (27%), and American Hispanic (15%). The median baseline CD4+ cell count was 262 cells per mm3 (range: 21 to 918 cells per mm3) and the median baseline plasma HIV-1 RNA was 4.89 log10 copies per mL (range: 2.6 to 6.99 log10 copies per mL).
The outcomes of randomized treatment are provided in Table 10.
Table 10. Outcomes of Randomized Treatment through Week 48 (CNA30021)|
a Subjects achieved and maintained confirmed HIV-1 RNA less than 50 copies per
mL (less than 400 copies per mL) through Week 48 (Roche AMPLICOR
Ultrasensitive HIV-1 MONITOR standard test version 1). | ||
|
Outcome |
Abacavir 600 mg |
Abacavir 300 mg |
|
Respondera |
64% (71%) |
65% (72%) |
|
Virologic failureb |
11% (5%) |
11% (5%) |
|
Discontinued due to adverse reactions |
13% |
11% |
|
Discontinued due to other reasonsc |
11% |
13% |
After 48 weeks of therapy, the median CD4+ cell count increases from baseline were 188 cells per mm3 in the group receiving abacavir 600 mg once daily and 200 cells per mm3 in the group receiving abacavir 300 mg twice daily. Through Week 48, 6 subjects (2%) in the group receiving abacavir 600 mg once daily (4 CDC classification C events and 2 deaths) and 10 subjects (3%) in the group receiving abacavir 300 mg twice daily (7 CDC classification C events and 3 deaths) experienced clinical disease progression. None of the deaths were attributed to trial medications.
14.2 Pediatric Trials
Therapy-Experienced Pediatric Subjects
CNA3006 was a randomized, double-blind trial comparing abacavir 8 mg per kg twice daily plus lamivudine 4 mg per kg twice daily plus zidovudine 180 mg per m2 twice daily versus lamivudine 4 mg per kg twice daily plus zidovudine 180 mg per m2 twice daily. Two hundred and five therapy-experienced pediatric subjects were enrolled: female (56%), white (17%), black (50%), Hispanic (30%), median age of 5.4 years, baseline CD4+ cell percent greater than 15% (median = 27%), and median baseline plasma HIV-1 RNA of 4.6 log10 copies per mL. Eighty percent and 55% of subjects had prior therapy with zidovudine and lamivudine, respectively, most often in combination. The median duration of prior nucleoside analogue therapy was 2 years. At 16 weeks, the proportion of subjects responding based on plasma HIV-1 RNA less than or equal to 400 copies per mL was significantly higher in subjects receiving abacavir plus lamivudine plus zidovudine compared with subjects receiving lamivudine plus zidovudine, 13% versus 2%, respectively. Median plasma HIV-1 RNA changes from baseline were -0.53 log10 copies per mL in the group receiving abacavir plus lamivudine plus zidovudine compared with -0.21 log10 copies per mL in the group receiving lamivudine plus zidovudine. Median CD4+ cell count increases from baseline were 69 cells per mm3 in the group receiving abacavir plus lamivudine plus zidovudine and 9 cells per mm3 in the group receiving lamivudine plus zidovudine.
Once-Daily Dosing
ARROW (COL105677) was a 5-year randomized, multicenter trial which evaluated multiple aspects of clinical management of HIV-1 infection in pediatric subjects. HIV-1–infected, treatment-naive subjects aged 3 months to 17 years were enrolled and treated with a first-line regimen containing abacavir and lamivudine, dosed twice daily according to World Health Organization recommendations. After a minimum of 36 weeks of treatment, subjects were given the option to participate in Randomization 3 of the ARROW trial, comparing the safety and efficacy of once-daily dosing with twice-daily dosing of abacavir and lamivudine, in combination with a third antiretroviral drug, for an additional 96 weeks. Of the 1,206 original ARROW subjects, 669 participated in Randomization 3. Virologic suppression was not a requirement for participation at baseline for Randomization 3 (following a minimum of 36 weeks of twice- daily treatment), 75% of subjects in the twice-daily cohort were virologically suppressed compared with 71% of subjects in the once-daily cohort.
The proportions of subjects with HIV-1 RNA less than 80 copies per mL through 96 weeks are shown in Table 11. The differences between virologic responses in the two treatment arms were comparable across baseline characteristics for gender and age.
Table 11. Virologic Outcome of Randomized Treatment at Week 96a (ARROW Randomization 3)|
a Analyses were based on the last observed viral load data within the Week 96
window. | ||
|
Outcome |
Abacavir plus |
Abacavir plus |
|
HIV-1 RNA <80 copies/mL****b |
70% 1% |
67% <1% |
SPL UNCLASSIFIED SECTION
(Front of Card)
** WARNING CARD**
Abacavir Tablets USP
Patients taking abacavir tablets may have a serious allergic reaction (hypersensitivity reaction) that can cause death. If you get a symptom from 2 or more of the following groups while taking abacavir tablets, call your healthcare provider right away to find out if you should stop taking this medicine.
|
Symptom(s) | |
|
** Group 1** |
** Fever** |
|
** Group 2** |
** Rash** |
|
** Group 3** |
** Nausea, vomiting, diarrhea, or abdominal (stomach area) pain** |
|
** Group 4** |
** Generally ill feeling, extreme tiredness, or achiness** |
|
** Group 5** |
** Shortness of breath, cough, or sore throat** |
Always carry this Warning Card with you to help recognize symptoms of this allergic reaction.
(Back of Card)
** WARNING CARD******
Abacavir Tablets USP
If you must stop treatment with abacavir tablets because you have had an allergic reaction to abacavir,NEVER take abacavir tablets or another abacavir-containing medicine (EPZICOM®, TRIUMEQ ®, or TRIZIVIR®) again. If you have an allergic reaction, dispose of any unused abacavir tablets. Ask your pharmacist how to properly dispose of medicines. If you take abacavir tablets or another abacavir-containing medicine again after you have had an allergic reaction,WITHIN HOURS you may getlife-threatening****symptoms that may includevery low blood pressure ordeath.
Please read the Medication Guide for additional information on abacavir tablets.
EPZICOM, TRIUMEQ, and TRIZIVIR are registered trademarks of the ViiV Healthcare group of companies.
Distributed by:
Aurobindo Pharma USA, Inc.
****279 Princeton-Hightstown Road
East Windsor, NJ 08520
Manufactured by:
Aurobindo Pharma Limited
Hyderabad-500 032, India
Revised: 12/2022