
IBANDRONATE SODIUM
{'content': {'@styleCode': 'bold', 'content': {'@styleCode': 'bold', 'content': [{'@styleCode': 'bold', '#text': 'IBANDRONATE SODIUM'}, {'@styleCode': 'bold', '#text': 'IBANDRONATE'}], 'br': [None, None, None], '#text': 'IBANDRONATE SODIUM TABLETS. These highlights do not include all the information needed to use \n TABLETS safely and effectively. See full prescribing information for \n SODIUM TABLETS.\n \n \nIBANDRONATE SODIUM tablets, for oral use\n \nInitial U.S. Approval: 2003'}}}
618e1aff-ccbb-4cee-93f1-fe04e33dacb7
HUMAN PRESCRIPTION DRUG LABEL
Sep 25, 2023
Golden State Medical Supply, Inc.
DUNS: 603184490
Products 1
Detailed information about drug products covered under this FDA approval, including NDC codes, dosage forms, ingredients, and administration routes.
IBANDRONATE SODIUM
Product Details
FDA regulatory identification and product classification information
FDA Identifiers
Product Classification
Product Specifications
INGREDIENTS (8)
Drug Labeling Information
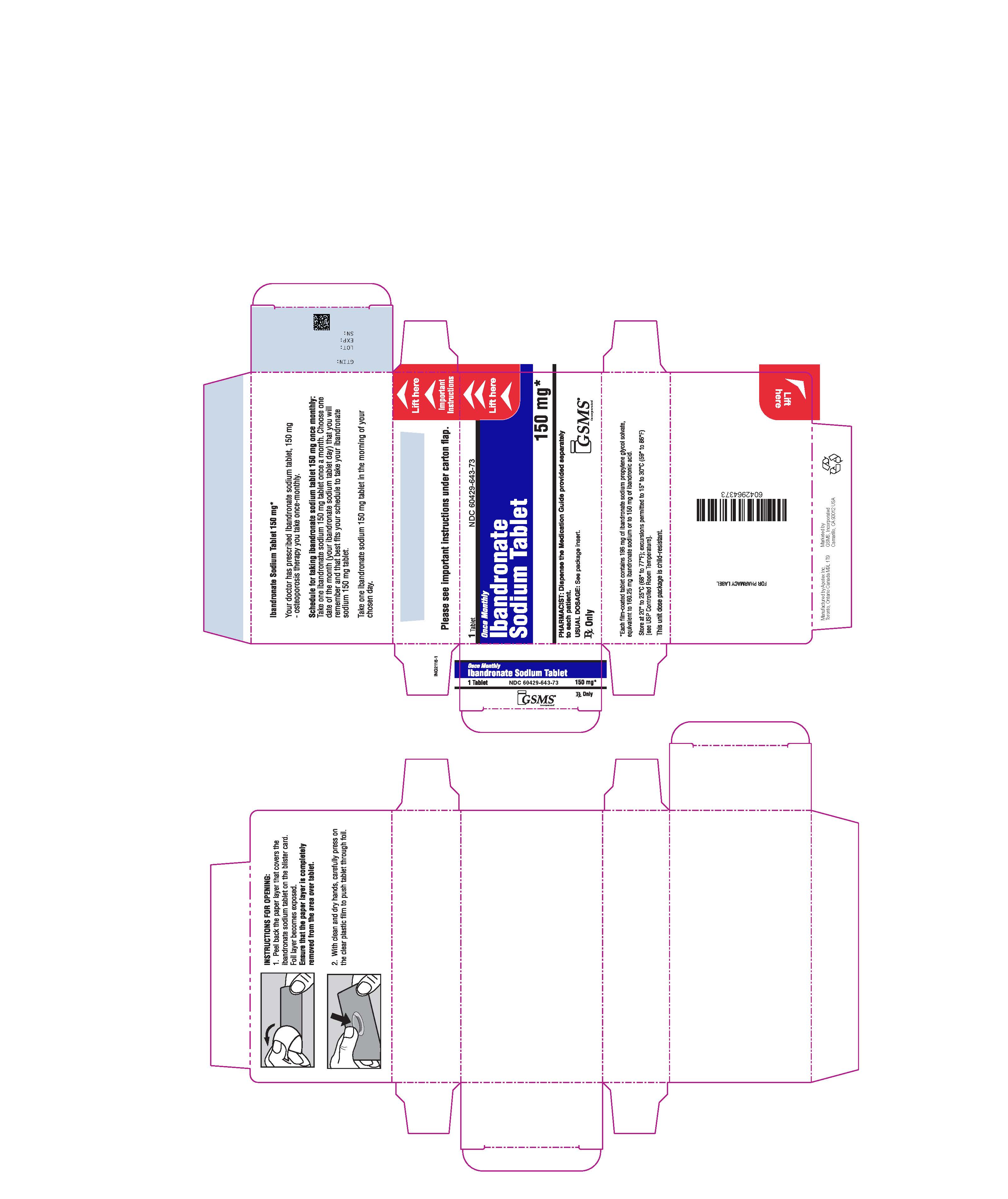
PACKAGE LABEL.PRINCIPAL DISPLAY PANEL
PRINCIPAL DISPLAY PANEL - 150 mg CARTON LABEL
NDC No. 60429-643-73
Ibandronate Sodium Tablet
Each tablet contains 196 mg of ibandronate sodium propylene glycol solvate equivalent to 160.25 mg ibandronate sodium or 150 mg of ibandronic acid.
150 mg
Rx Only
1 tablet
DRUG INTERACTIONS SECTION
7 DRUG INTERACTIONS
7.1 Calcium Supplements/Antacids
Products containing calcium and other multivalent cations (such as aluminum, magnesium, iron) are likely to interfere with absorption of ibandronate sodium tablets. Therefore, instruct patients to take ibandronate sodium tablets at least 60 minutes before any oral medications, including medications containing multivalent cations (such as antacids, supplements or vitamins). Also, patients should wait at least 60 minutes after dosing before taking any other oral medications ( seeDOSAGE AND ADMINISTRATION [2.3]).
7.2 Aspirin/Nonsteroidal Anti-Inflammatory Drugs (NSAIDs)
Because aspirin, NSAIDs, and bisphosphonates are all associated with gastrointestinal irritation, caution should be exercised in the concomitant use of aspirin or NSAIDs with ibandronate sodium tablets.
7.3 H2 Blockers
In healthy volunteers, co-administration with ranitidine resulted in a 20% increased bioavailability of ibandronate, which was not considered to be clinically relevant (see CLINICAL PHARMACOLOGY [12.3]) .
7.4 Drug/Laboratory Test Interactions
Bisphosphonates are known to interfere with the use of bone-imaging agents. Specific studies with ibandronate have not been performed.
- Calcium supplements, antacids and some oral medications may interfere with absorption of ibandronate. Do not take within 60 minutes of dosing ( 7.1)
- Use caution when co-prescribing aspirin/nonsteroidal anti-inflammatory drugs that may worsen gastrointestinal irritation. ( 7.2)
CLINICAL PHARMACOLOGY SECTION
12 CLINICAL PHARMACOLOGY
12.1 Mechanism of Action
The action of ibandronate on bone tissue is based on its affinity for hydroxyapatite, which is part of the mineral matrix of bone. Ibandronate inhibits osteoclast activity and reduces bone resorption and turnover. In postmenopausal women, it reduces the elevated rate of bone turnover, leading to, on average, a net gain in bone mass.
12.2 Pharmacodynamics
Osteoporosis is characterized by decreased bone mass and increased fracture risk, most commonly at the spine, hip, and wrist. The diagnosis can be confirmed by a finding of low bone mass, evidence of fracture on x-ray, a history of osteoporotic fracture, or height loss or kyphosis indicative of vertebral fracture. While osteoporosis occurs in both men and women, it is most common among women following menopause. In healthy humans, bone formation and resorption are closely linked; old bone is resorbed and replaced by newly formed bone. In postmenopausal osteoporosis, bone resorption exceeds bone formation, leading to bone loss and increased risk of fracture. After menopause, the risk of fractures of the spine and hip increases; approximately 40% of 50-year-old women will experience an osteoporosis-related fracture during their remaining lifetimes.
Ibandronate sodium tablets produced biochemical changes indicative of dose- dependent inhibition of bone resorption, including decreases of biochemical markers of bone collagen degradation (such as deoxypyridinoline, and cross- linked C-telopeptide of Type I collagen) in the daily dose range of 0.25 mg to 5 mg and once-monthly doses from 100 mg to 150 mg in postmenopausal women.
Treatment with 2.5 mg daily ibandronate sodium tablets resulted in decreases in biochemical markers of bone turnover, including urinary C-terminal telopeptide of Type I collagen (uCTX) and serum osteocalcin, to levels similar to those in premenopausal women. Changes in markers of bone formation were observed later than changes in resorption markers, as expected, due to the coupled nature of bone resorption and formation. Treatment with 2.5 mg daily ibandronate sodium tablets decreased levels of uCTX within 1 month of starting treatment and decreased levels of osteocalcin within 3 months. Bone turnover markers reached a nadir of approximately 64% below baseline values by 6 months of treatment and remained stable with continued treatment for up to 3 years. Following treatment discontinuation, there is a return to pretreatment baseline rates of elevated bone resorption associated with postmenopausal osteoporosis.
In a 1-year, study comparing once-monthly vs. once-daily oral dosing regimens, the median decrease from baseline in serum CTX values was -76% for patients treated with the 150 mg once-monthly regimen and -67% for patients treated with the 2.5 mg daily regimen. In a 1-year, prevention study comparing ibandronate sodium tablets 150 mg once-monthly to placebo, the median placebo- subtracted decrease in sCTX was -49.8%.
12.3 Pharmacokinetics
Absorption
The absorption of oral ibandronate occurs in the upper gastrointestinal tract. Plasma concentrations increase in a dose-linear manner up to 50 mg oral intake and increases nonlinearly above this dose.
Following oral dosing, the time to maximum observed plasma ibandronate concentrations ranged from 0.5 to 2 hours (median 1 hour) in fasted healthy postmenopausal women. The mean oral bioavailability of 2.5 mg ibandronate was about 0.6% compared to intravenous dosing. The extent of absorption is impaired by food or beverages (other than plain water). The oral bioavailability of ibandronate is reduced by about 90% when ibandronate sodium tablets are administered concomitantly with a standard breakfast in comparison with bioavailability observed in fasted subjects. There is no meaningful reduction in bioavailability when ibandronate is taken at least 60 minutes before a meal. However, both bioavailability and the effect on bone mineral density (BMD) are reduced when food or beverages are taken less than 60 minutes following an ibandronate dose.
Distribution
After absorption, ibandronate either rapidly binds to bone or is excreted into urine. In humans, the apparent terminal volume of distribution is at least 90 L, and the amount of dose removed from the circulation via the bone is estimated to be 40% to 50% of the circulating dose. In vitroprotein binding in human serum was 99.5% to 90.9% over an ibandronate concentration range of 2 to 10 ng/mL in one study and approximately 85.7% over a concentration range of 0.5 to 10 ng/mL in another study.
Metabolism
Ibandronate does not undergo hepatic metabolism and does not inhibit the hepatic cytochrome P450 system. Ibandronate is eliminated by renal excretion. Based on a rat study, the ibandronate secretory pathway does not appear to include known acidic or basic transport systems involved in the excretion of other drugs. There is no evidence that ibandronate is metabolized in humans.
Elimination
The portion of ibandronate that is not removed from the circulation via bone absorption is eliminated unchanged by the kidney (approximately 50% to 60% of the absorbed dose). Unabsorbed ibandronate is eliminated unchanged in the feces.
The plasma elimination of ibandronate is multiphasic. Its renal clearance and distribution into bone accounts for a rapid and early decline in plasma concentrations, reaching 10% of the C maxwithin 3 or 8 hours after intravenous or oral administration, respectively. This is followed by a slower clearance phase as ibandronate redistributes back into the blood from bone. The observed apparent terminal half-life for ibandronate is generally dependent on the dose studied and on assay sensitivity. The observed apparent terminal half-life for the 150 mg ibandronate tablet upon oral administration to healthy postmenopausal women ranges from 37 to 157 hours.
Total clearance of ibandronate is low, with average values in the range 84 to 160 mL/min. Renal clearance (about 60 mL/min in healthy postmenopausal females) accounts for 50% to 60% of total clearance and is related to creatinine clearance. The difference between the apparent total and renal clearances likely reflects bone uptake of the drug.
Specific Populations
Pediatrics
The pharmacokinetics of ibandronate has not been studied in patients less than 18 years of age.
Geriatric
Because ibandronate is not known to be metabolized, the only difference in ibandronate elimination for geriatric patients versus younger patients is expected to relate to progressive age-related changes in renal function.
Gender
The bioavailability and pharmacokinetics of ibandronate are similar in both men and women.
Race
Pharmacokinetic differences due to race have not been studied.
Renal Impairment
Renal clearance of ibandronate in patients with various degrees of renal impairment is linearly related to creatinine clearance (CLcr).
Following a single dose of 0.5 mg ibandronate by intravenous administration, patients with CLcr 40 to 70 mL/min had 55% higher exposure (AUC ∞) than the exposure observed in subjects with CLcr greater than 90 mL/min. Patients with CLcr less than 30 mL/min had more than a two-fold increase in exposure compared to the exposure for healthy subjects (see DOSAGE AND ADMINISTRATION [2.4]) .
Hepatic Impairment
No studies have been performed to assess the pharmacokinetics of ibandronate in patients with hepatic impairment because ibandronate is not metabolized in the human liver.
Drug Interaction Studies
Products containing calcium and other multivalent cations (such as aluminum, magnesium, iron), including milk, food, and antacids are likely to interfere with absorption of ibandronate, which is consistent with findings in animal studies.
H2 Blockers
A pharmacokinetic interaction study in healthy volunteers demonstrated that 75 mg ranitidine (25 mg injected intravenously 90 and 15 minutes before and 30 minutes after ibandronate administration) increased the oral bioavailability of 10 mg ibandronate by about 20%. This degree of increase is not considered to be clinically relevant.
HOW SUPPLIED SECTION
16 HOW SUPPLIED/STORAGE AND HANDLING
16.1 How Supplied
Ibandronate sodium 150-mg tablets are supplied as white to off-white, oval, biconvex, film-coated tablets, engraved with "APO" on one side and "IBA150" on the other side. They are supplied as follows:
Packaged in bundles of 3 boxes, each box containing 1 blister strip of 1 tablet each (NDC 60429-643-73).
16.2 Storage and Handling
Store at 20° to 25°C (68° to 77°F); excursions permitted between 15° and 30°C (59° and 86°F) [see USP Controlled Room Temperature].
Dispense in a tight container [see USP].