
Daytrana
These highlights do not include all the information needed to use DAYTRANA safely and effectively. See full prescribing information for DAYTRANA. DAYTRANA (methylphenidate transdermal system), CII Initial U.S. Approval: 2006
2c312c31-3198-4775-91ab-294e0b4b9e7f
HUMAN PRESCRIPTION DRUG LABEL
Oct 18, 2023
Noven Therapeutics, LLC
DUNS: 166888268
Products 4
Detailed information about drug products covered under this FDA approval, including NDC codes, dosage forms, ingredients, and administration routes.
methylphenidate
Product Details
FDA regulatory identification and product classification information
FDA Identifiers
Product Classification
Product Specifications
INGREDIENTS (1)
methylphenidate
Product Details
FDA regulatory identification and product classification information
FDA Identifiers
Product Classification
Product Specifications
INGREDIENTS (1)
methylphenidate
Product Details
FDA regulatory identification and product classification information
FDA Identifiers
Product Classification
Product Specifications
INGREDIENTS (1)
methylphenidate
Product Details
FDA regulatory identification and product classification information
FDA Identifiers
Product Classification
Product Specifications
INGREDIENTS (1)
Drug Labeling Information
PACKAGE LABEL.PRINCIPAL DISPLAY PANEL
PRINCIPAL DISPLAY PANEL - NDC 68968-5555-3 - 30 mg 30 Count Carton

NDC 68968-5555-3
Daytrana**®**
(methylphenidate transdermal system)
Delivers 30 mg over 9 hours
(3.3 mg/hr)
Patch should be worn for approximately 9 hours
Contains: 30 Patches
CII
Rx only
Noven Therapeutics, LLC
Once the tray is opened, use contents within 2 months.
Manufactured for Noven Therapeutics, LLC, Miami, FL 33186
By Noven Pharmaceuticals, Inc.
©2007, 2010 Noven Pharmaceuticals, Inc.
1-877-567-7857
Pharmacists: Enclosed Medication Guide to be dispensed to each patient.
302191-8 Rev. 9/2016
Each patch contains 82.5 mg of methylphenidate.
Active ingredient release is limited; please see recommended dosing in patient instructions.
Inactive components: acrylic adhesive, coextruded backing film, polyester release liner and silicone adhesive.
For transdermal use only (applied only to skin).
Keep all patches within provided containers and dispense one patch daily. Apply immediately upon removal from pouch.
Do not store unpouched. Do not store patches in refrigerators or freezers.
Store at 25°C (77°F); excursions permitted to 15-30°C (59-86°F) [see USP controlled Room Temperature]
Important: Keep out of the reach of children.
It is important that this product be disposed of properly. See patient instructions for disposal information.
Dosage and Administration: See package insert.
|
Date of Patch Application MM/DD/YYYY |
Time Applied |
Time Removed |
Application Side |
Disposal Method |
Date of Patch Application MM/DD/YYYY |
Time Applied |
Time Removed |
Application Side |
Disposal Method |
|
_AM _PM |
_AM _PM |
_Right _Left |
_Fold & flush _ Fold & trash |
_AM _PM |
_AM _PM |
_Right _Left |
_Fold & flush _ Fold & trash | ||
|
_AM _PM |
_AM _PM |
_Right _Left |
_Fold & flush _ Fold & trash |
_AM _PM |
_AM _PM |
_Right _Left |
_Fold & flush _ Fold & trash | ||
|
_AM _PM |
_AM _PM |
_Right _Left |
_Fold & flush _ Fold & trash |
_AM _PM |
_AM _PM |
_Right _Left |
_Fold & flush _ Fold & trash | ||
|
_AM _PM |
_AM _PM |
_Right _Left |
_Fold & flush _ Fold & trash |
_AM _PM |
_AM _PM |
_Right _Left |
_Fold & flush _ Fold & trash | ||
|
_AM _PM |
_AM _PM |
_Right _Left |
_Fold & flush _ Fold & trash |
_AM _PM |
_AM _PM |
_Right _Left |
_Fold & flush _ Fold & trash | ||
|
_AM _PM |
_AM _PM |
_Right _Left |
_Fold & flush _ Fold & trash |
_AM _PM |
_AM _PM |
_Right _Left |
_Fold & flush _ Fold & trash | ||
|
_AM _PM |
_AM _PM |
_Right _Left |
_Fold & flush _ Fold & trash |
_AM _PM |
_AM _PM |
_Right _Left |
_Fold & flush _ Fold & trash | ||
|
_AM _PM |
_AM _PM |
_Right _Left |
_Fold & flush _ Fold & trash |
_AM _PM |
_AM _PM |
_Right _Left |
_Fold & flush _ Fold & trash | ||
|
_AM _PM |
_AM _PM |
_Right _Left |
_Fold & flush _ Fold & trash |
_AM _PM |
_AM _PM |
_Right _Left |
_Fold & flush _ Fold & trash | ||
|
_AM _PM |
_AM _PM |
_Right _Left |
_Fold & flush _ Fold & trash |
_AM _PM |
_AM _PM |
_Right _Left |
_Fold & flush _ Fold & trash | ||
|
_AM _PM |
_AM _PM |
_Right _Left |
_Fold & flush _ Fold & trash |
_AM _PM |
_AM _PM |
_Right _Left |
_Fold & flush _ Fold & trash | ||
|
_AM _PM |
_AM _PM |
_Right _Left |
_Fold & flush _ Fold & trash |
_AM _PM |
_AM _PM |
_Right _Left |
_Fold & flush _ Fold & trash | ||
|
_AM _PM |
_AM _PM |
_Right _Left |
_Fold & flush _ Fold & trash |
_AM _PM |
_AM _PM |
_Right _Left |
_Fold & flush _ Fold & trash | ||
|
_AM _PM |
_AM _PM |
_Right _Left |
_Fold & flush _ Fold & trash |
_AM _PM |
_AM _PM |
_Right _Left |
_Fold & flush _ Fold & trash | ||
|
_AM _PM |
_AM _PM |
_Right _Left |
_Fold & flush _ Fold & trash |
_AM _PM |
_AM _PM |
_Right _Left |
_Fold & flush _ Fold & trash |
WARNINGS AND PRECAUTIONS SECTION
5 WARNINGS AND PRECAUTIONS
5.1 Abuse, Misuse, and Addiction
DAYTRANA has a high potential for abuse and misuse. The use of DAYTRANA exposes individuals to the risks of abuse and misuse, which can lead to the development of a substance use disorder, including addiction. DAYTRANA can be diverted for non-medical use into illicit channels or distribution [see Drug Abuse and Dependence (9.2)]. Misuse and abuse of CNS stimulants, including DAYTRANA, can result in overdose and death [see Overdosage (10)], and this risk is increased with higher doses or unapproved methods of administration, such as snorting or injection.
Before prescribing DAYTRANA, assess each patient’s risk for abuse, misuse, and addiction. Educate patients and their caregivers or families about these risks. Advise patients to store DAYTRANA in a safe place, preferably locked, and instruct patients to not give DAYTRANA to anyone else. Throughout DAYTRANA treatment, reassess each patient’s risk of abuse, misuse, and addiction and frequently monitor for signs and symptoms of abuse, misuse, and addiction.
DAYTRANA has special disposal instructions. Instruct patients to find a take back location to dispose of unused or expired DAYTRANA. If a take back program is unavailable, instruct them to:
- Remove DAYTRANA from its pouch, separate it from its liner, fold it in half with the adhesive sides touching each other, and immediately flush the used transdermal system down the toilet, and
- Place the pouch and liner in a container, close the container, and throw out the container in the trash (advise patients not to flush the pouch and liner down the toilet).
5.2 Risks to Patients with Serious Cardiac Disease
Sudden death has been reported in patients with structural cardiac abnormalities or other serious cardiac disease who were treated with CNS stimulants at the recommended ADHD dosage.
Avoid DAYTRANA use in patients with known structural cardiac abnormalities, cardiomyopathy, serious cardiac arrhythmia, coronary artery disease, or other serious cardiac disease.
5.3 Increased Blood Pressure and Heart Rate
CNS stimulants may cause an increase in blood pressure (mean increase approximately 2 to 4 mmHg) and heart rate (mean increase approximately 3 to 6 bpm). Some patients may have larger increases.
Monitor all DAYTRANA-treated patients for hypertension and tachycardia.
5.4 Psychiatric Adverse Reactions
Exacerbation of Pre-Existing Psychosis
CNS stimulants may exacerbate symptoms of behavior disturbance and thought disorder in patients with a pre-existing psychotic disorder.
Induction of a Manic Episode in Patients with Bipolar Disease
CNS stimulants may induce a manic or mixed episode in patients. Prior to initiating DAYTRANA treatment, screen patients for risk factors for developing a manic episode (e.g., comorbid or history of depressive symptoms or a family history of suicide, bipolar disorder, or depression).
New Psychotic or Manic Symptoms
CNS stimulants, at the recommended dosages, may cause psychotic or manic symptoms, (e.g., hallucinations, delusional thinking, or mania) in patients without a prior history of psychotic illness or mania. In a pooled analysis of multiple short-term, placebo-controlled studies of CNS stimulants, psychotic or manic symptoms occurred in approximately 0.1% of CNS stimulant-treated patients compared with 0% of placebo-treated patients. If such symptoms occur, consider discontinuing DAYTRANA.
5.5 Seizures
There is some clinical evidence that stimulants may lower the convulsive threshold in patients with prior history of seizures, in patients with prior EEG abnormalities in absence of seizures, and, very rarely, in patients without a history of seizures and no prior EEG evidence of seizures. In the presence of seizures, the drug should be discontinued.
5.6 Priapism
Prolonged and painful erections, sometimes requiring surgical intervention, have been reported with methylphenidate use in both adult and pediatric male patients. Although priapism was not reported with methylphenidate initiation, it developed after some time on the methylphenidate, often subsequent to an increase in dosage. Priapism also occurred during methylphenidate withdrawal (drug holidays or during discontinuation).
DAYTRANA-treated patients who develop abnormally sustained or frequent and painful erections should seek immediate medical attention.
5.7 Peripheral Vasculopathy, including Raynaud’s phenomenon
Stimulant medications, including DAYTRANA, used to treat ADHD are associated with peripheral vasculopathy, including Raynaud’s phenomenon. Signs and symptoms are usually intermittent and mild; however, sequelae have included digital ulceration and/or soft tissue breakdown. Effects of peripheral vasculopathy, including Raynaud’s phenomenon, were observed in post-marketing reports and at therapeutic dosages of CNS stimulants in all age groups throughout the course of treatment. Signs and symptoms generally improved after dosage reduction or discontinuation of the CNS stimulant.
Careful observation for digital changes is necessary during DAYTRANA treatment. Further clinical evaluation (e.g., rheumatology referral) may be appropriate for DAYTRANA-treated patients who develop signs or symptoms of peripheral vasculopathy.
5.8 Long-Term Suppression of Growth in Pediatric Patients
CNS stimulants have been associated with weight loss and slowing of growth rate in pediatric patients.
Careful follow-up of weight and height in children ages 7 to 10 years who were randomized to either methylphenidate or non-medication treatment groups over 14 months, as well as in naturalistic subgroups of newly methylphenidate- treated and non-medication treated children over 36 months (to the ages of 10 to 13 years), suggests that pediatric patients who received methylphenidate for 7 days per week throughout the year had a temporary slowing in growth rate (on average, a total of about 2 cm less growth in height and 2.7 kg less growth in weight over 3 years), without evidence of growth rebound during this development period.
Closely monitor growth (weight and height) in DAYTRANA-treated pediatric patients. Pediatric patients not growing or gaining height or weight as expected may need to have their treatment interrupted.
5.9 Chemical Leukoderma
DAYTRANA use may result in a persistent loss of skin pigmentation at and around the application site. Loss of pigmentation, in some cases, has been reported at other sites distant from the application site. Chemical leukoderma can mimic the appearance of vitiligo, particularly when the loss of skin pigmentation involves areas distant from the application site. Individuals with a history of vitiligo and/or a family history of vitiligo may be more at risk. Skin depigmentation may persist even after DAYTRANA use is discontinued. Monitor for signs of skin depigmentation, and advise patients to immediately inform their healthcare provider if changes in skin pigmentation occur. Discontinue use of the DAYTRANA in patients with chemical leukoderma.
5.10 Contact Sensitization
In an open-label study of 305 subjects conducted to characterize dermal reactions in children with ADHD treated with DAYTRANA using a 9-hour wear time, one subject (0.3%) was confirmed by patch testing to be sensitized to methylphenidate (allergic contact dermatitis). This subject experienced erythema and edema at DAYTRANA application sites with concurrent urticarial lesions on the abdomen and legs resulting in treatment discontinuation. This subject was not transitioned to oral methylphenidate.
Use of DAYTRANA may lead to contact sensitization. DAYTRANA should be discontinued if contact sensitization is suspected. Erythema is commonly seen with use of DAYTRANA and is not by itself an indication of sensitization. However, contact sensitization should be suspected if erythema is accompanied by evidence of a more intense local reaction (edema, papules, vesicles) that does not significantly improve within 48 hours or spreads beyond the transdermal system site. Confirmation of a diagnosis of contact sensitization (allergic contact dermatitis) may require further diagnostic testing.
Patients sensitized from use of DAYTRANA, as evidenced by development of an allergic contact dermatitis, may develop systemic sensitization or other systemic reactions if methylphenidate-containing products are taken via other routes, e.g., orally. Manifestations of systemic sensitization may include a flare-up of previous dermatitis or of prior positive patch-test sites, or generalized skin eruptions in previously unaffected skin. Other systemic reactions may include headache, fever, malaise, arthralgia, diarrhea, or vomiting. No cases of systemic sensitization have been observed in clinical trials of DAYTRANA.
Patients who develop contact sensitization to DAYTRANA and require oral treatment with methylphenidate should be initiated on oral medication under close medical supervision. It is possible that some patients sensitized to methylphenidate by exposure to DAYTRANA may not be able to take methylphenidate in any form.
5.11 Patients Using External Heat
Patients should be advised to avoid exposing the DAYTRANA application site to direct external heat sources, such as hair dryers, heating pads, electric blankets, heated water beds, etc., while wearing the transdermal system. When heat is applied to DAYTRANA after application, both the rate and extent of absorption are significantly increased. The temperature-dependent increase in methylphenidate absorption can be greater than 2-fold [see Clinical Pharmacology (12.3)]. This increased absorption can be clinically significant and can result in overdose of methylphenidate [see Overdosage (10)].
5.12 Hematologic Monitoring
Periodic CBC, differential, and platelet counts are advised during prolonged therapy.
5.13 Acute Angle Closure Glaucoma
There have been reports of angle closure glaucoma associated with methylphenidate treatment.
Although the mechanism is not clear, DAYTRANA-treated patients considered at risk for acute angle closure glaucoma (e.g., patients with significant hyperopia) should be evaluated by an ophthalmologist.
5.14 Increased Intraocular Pressure and Glaucoma
There have been reports of an elevation of intraocular pressure (IOP) associated with methylphenidate treatment [see Adverse Reactions (6.2)] .
Prescribe DAYTRANA to patients with open-angle glaucoma or abnormally increased IOP only if the benefit of treatment is considered to outweigh the risk. Closely monitor DAYTRANA-treated patients with a history of abnormally increased IOP or open angle glaucoma.
5.15 Motor and Verbal Tics, and Worsening of Tourette’s Syndrome
CNS stimulants, including methylphenidate, have been associated with the onset or exacerbation of motor and verbal tics. Worsening of Tourette’s syndrome has also been reported [see Adverse Reactions (6.2)] .
Before initiating DAYTRANA, assess the family history and clinically evaluate patients for tics or Tourette’s syndrome. Regularly monitor DAYTRANA-treated patients for the emergence or worsening of tics or Tourette’s syndrome, and discontinue treatment if clinically appropriate.
- Risks to Patients with Serious Cardiac Disease: Avoid use in patients with known structural cardiac abnormalities, cardiomyopathy, serious cardiac arrhythmias, coronary artery disease, or other serious cardiac disease. (5.2)
- Increased Blood Pressure and Heart Rate: Monitor blood pressure and pulse. (5.3)
- Psychiatric Adverse Reactions: Prior to initiating DAYTRANA, screen patients for risk factors for developing a manic episode. If new psychotic or manic symptoms occur, consider discontinuing DAYTRANA. (5.4)
- Seizures: Stimulants may lower the convulsive threshold. Discontinue in the presence of seizures. (5.5)
- Priapism: If abnormally sustained or frequent and painful erections occur, patients should seek immediate medical attention. (5.6)
- Peripheral Vasculopathy, including Raynaud’s phenomenon: Careful observation for digital changes is necessary during DAYTRANA treatment. Further clinical evaluation (e.g., rheumatology referral) may be appropriate for patients who develop signs or symptoms of peripheral vasculopathy. (5.7)
- Long-Term Suppression of Growth in Pediatric Patients: Closely monitor (height and weight) in pediatric patients. Pediatric patients not growing or gaining height or weight as expected may need to have their treatment interrupted. (5.8)
- Chemical Leukoderma: DAYTRANA use may result in a persistent loss of skin pigmentation at and around the application site. Loss of pigmentation, in some cases, has been reported at other sites distant from the application site. Monitor for signs of skin depigmentation. Discontinue DAYTRANA if it occurs. (5.9)
- Contact Sensitization: Use of DAYTRANA may lead to contact sensitization. Treatment should be discontinued if contact sensitization is suspected. Erythema is commonly seen with use of DAYTRANA and is not by itself an indication of sensitization. However, contact sensitization should be suspected if erythema is accompanied by evidence of a more intense local reaction (edema, papules, vesicles) that does not significantly improve within 48 hours or spreads beyond the transdermal system site. (5.10)
- External Heat: Patients should be advised to avoid exposing the DAYTRANA application site to direct external heat sources. When heat is applied to DAYTRANA after application, both the rate and extent of absorption are significantly increased. (5.11)
- Hematologic monitoring: Periodic CBC, differential, and platelet counts are advised during prolonged therapy. (5.12)
- Acute Angle Closure Glaucoma: DAYTRANA-treated patients considered at risk for acute angle closure glaucoma (e.g., patients with significant hyperopia) should be evaluated by an ophthalmologist. (5.13)
- Increased Intraocular Pressure (IOP) and Glaucoma: Prescribe DAYTRANA to patients with open-angle glaucoma or abnormally increased IOP only if the benefit of treatment is considered to outweigh the risk. Closely monitor patients with a history of increased IOP or open angle glaucoma. (5.14)
- Motor and Verbal Tics and Worsening of Tourette’s Syndrome: Before initiating DAYTRANA, assess the family history and clinically evaluate patients for tics or Tourette’s syndrome. Regularly monitor patients for the emergence or worsening of tics or Tourette’s syndrome. Discontinue treatment if clinically appropriate. (5.15)
ADVERSE REACTIONS SECTION
6 ADVERSE REACTIONS
Detailed information on serious and adverse reactions of particular importance is provided in the Boxed Warning and Warnings and Precautions (5) sections:
- Abuse, Misuse, and Addiction [see Boxed Warning]
- Hypersensitivity to Methylphenidate [see Contraindications (4.1)]
- Monoamine Oxidase Inhibitors [see Contraindications (4.2) and Drug Interactions (7.1)]
- Risks to Patients with Serious Cardiac Disease [see Warnings and Precautions (5.2)]
- Increased Blood Pressure and Heart Rate [see Warnings and Precautions (5.3)]
- Psychiatric Adverse Reactions [see Warnings and Precautions (5.4)]
- Seizures [see Warnings and Precautions (5.5)]
- Priapism [see Warnings and Precautions (5.6)]
- Peripheral Vasculopathy [see Warnings and Precautions (5.7)]
- Long-Term Suppression of Growth in Pediatric Patients [see Warnings and Precautions (5.8)]
- Chemical Leukoderma [see Warnings and Precautions (5.9)]
- Contact Sensitization [see Warnings and Precautions (5.10)]
- External Heat [see Warnings and Precautions (5.11)]
- Hematologic Monitoring [see Warnings and Precautions (5.12)]
- Acute Angle Closure Glaucoma [see Warnings and Precautions (5.13)]
- Increased Intraocular Pressure and Glaucoma [see Warnings and Precautions (5.14)]
- Motor and Verbal Tics, and Worsening of Tourette’s Syndrome [see Warnings and Precautions (5.15)]
6.1 Clinical Trials Experience
Because clinical trials are conducted under widely varying conditions, adverse reactions rates observed in the clinical trials of a drug cannot be directly compared to rates in the clinical trials of another drug and may not reflect the rates observed in practice.
The most commonly reported (frequency ≥ 5% and twice the rate of placebo) adverse reactions in a controlled trial in children aged 6-12 included appetite decreased, insomnia, nausea, vomiting, weight decreased, tic, affect lability, and anorexia. The most commonly reported (frequency ≥ 5% and twice the rate of placebo) adverse reactions in a controlled trial in adolescents aged 13-17 were appetite decreased, nausea, insomnia, weight decreased, dizziness, abdominal pain, and anorexia [see Adverse Reactions (6.1)].
The most common (≥ 2% of subjects) adverse reaction associated with discontinuations in double-blind clinical trials in children or adolescents was application site reactions [see Adverse Reactions (6.1)].
The overall DAYTRANA development program included exposure to DAYTRANA in a total of 2,152 participants in clinical trials, including 1,529 children aged 6-12, 223 adolescents aged 13-17, and 400 adults. The 1,752 child and adolescent subjects aged 6-17 years were evaluated in 10 controlled clinical studies, 7 open-label clinical studies, and 5 clinical pharmacology studies. In a combined studies pool of children using DAYTRANA with a wear time of 9 hours, 212 subjects were exposed for ≥ 6 months and 115 were exposed for ≥ 1 year; 85 adolescents have been exposed for ≥ 6 months. Most patients studied were exposed to DAYTRANA transdermal system sizes of 12.5 cm2, 18.75 cm2, 25 cm2 or 37.5 cm2, with a wear time of 9 hours.
In the data presented below, the adverse reactions reported during exposure were obtained primarily by general inquiry at each visit, and were recorded by the clinical investigators using terminology of their own choosing. Consequently, it is not possible to provide a meaningful estimate of the proportion of individuals experiencing adverse reactions without first grouping similar types of events into a smaller number of standardized event categories.
Adverse Reactions in Clinical Studies with Discontinuation of Treatment
In a 7-week double-blind, parallel-group, placebo-controlled study in children with ADHD conducted in the outpatient setting, 7.1% (7/98) of patients treated with DAYTRANA discontinued due to adverse events compared with 1.2% (1/85) receiving placebo. The most commonly reported (≥ 1% and twice the rate of placebo) adverse reactions leading to discontinuation in the DAYTRANA group were application site reaction (2%), tics (1%), headache (1%), and irritability (1%).
In a 7-week double-blind, parallel-group, placebo-controlled study in adolescents with ADHD conducted in the outpatient setting, 5.5% (8/145) of patients treated with DAYTRANA discontinued due to adverse reactions compared with 2.8% (2/72) receiving placebo. The most commonly reported adverse reactions leading to discontinuation in the DAYTRANA group were application site reaction (2%) and decreased appetite/anorexia (1.4%).
Commonly Observed Adverse Reactions in Double-Blind, Placebo-Controlled Trials
Skin Irritation and Application Site Reactions
DAYTRANA is a dermal irritant. In addition to the most commonly reported adverse reactions presented in Table 2, the majority of subjects in those studies had minimal to definite skin erythema at the transdermal system application site. This erythema generally caused no or minimal discomfort and did not usually interfere with therapy or result in discontinuation from treatment. Erythema is not by itself a manifestation of contact sensitization. However, contact sensitization should be suspected if erythema is accompanied by evidence of a more intense local reaction (edema, papules, vesicles) that does not significantly improve within 48 hours or spreads beyond the transdermal system site [see Warnings and Precautions (5.10)].
Most Commonly Reported Adverse Reactions
Table 2 lists treatment-emergent adverse reactions reported in ≥ 1% DAYTRANA- treated children or adolescents with ADHD in two 7 week double-blind, parallel-group, placebo-controlled studies conducted in the outpatient setting. Overall, in these studies, 75.5% of children and 78.6% of adolescents experienced at least 1 adverse event.
| ||||
|
Table 2 Number (%) of Subjects with Commonly Reported Adverse Reactions (≥ 1% in the DAYTRANA Group) in 7-Week Placebo-controlled Studies in Either Children or Adolescents - Safety Population | ||||
|
Adolescents |
Children | |||
|
System Organ Class |
Placebo |
DAYTRANA |
Placebo |
DAYTRANA |
|
Cardiac Disorders | ||||
|
Tachycardia |
0 (0) |
1 (0.7) |
0 (0) |
1 (1.0) |
|
Gastrointestinal disorders | ||||
|
Abdominal pain |
0 (0) |
7 (4.8) |
5 (5.9) |
7 (7.1) |
|
Nausea |
2 (2.8) |
14 (9.7) |
2 (2.4) |
12 (12.2) |
|
Vomiting |
1 (1.4) |
5 (3.4) |
4 (4.7) |
10 (10.2) |
|
Investigations | ||||
|
Weight decreased |
1 (1.4) |
8 (5.5) |
0 (0) |
9 (9.2) |
|
Metabolism and nutrition disorders | ||||
|
Anorexia |
1 (1.4) |
7 (4.8) |
1 (1.2) |
5 (5.1) |
|
Decreased appetite |
1 (1.4) |
37 (25.5) |
4 (4.7) |
25 (25.5) |
|
Nervous system disorders | ||||
|
Dizziness |
1 (1.4) |
8 (5.5) |
1 (1.2) |
0 (0) |
|
Headache |
9 (12.5) |
18 (12.4) |
10 (11.8) |
15 (15.3) |
|
Psychiatric disorders | ||||
|
Affect lability |
1 (1.4) |
0 (0) |
0 (0) |
6 (6.1)* |
|
Insomnia |
2 (2.8) |
9 (6.2) |
4 (4.7) |
13 (13.3) |
|
Irritability |
5 (6.9) |
16 (11) |
4 (4.7) |
7 (7.1) |
|
Tic |
0 (0) |
0 (0) |
0 (0) |
7 (7.1) |
Adverse Reactions in Clinical Studies with the Long-Term Use of DAYTRANA
In a long-term open-label study of up to 12 months duration in 326 children wearing DAYTRANA 9 hours daily, the most common (≥ 10%) adverse reactions were decreased appetite, headache, and weight decreased. A total of 30 subjects (9.2%) were withdrawn from the study due to adverse events and 22 additional subjects (6.7%) discontinued treatment as the result of an application site reaction. Other than application site reactions, affect lability (5 subjects, 1.5%) was the only additional adverse reaction leading to discontinuation reported with a frequency of greater than 1%.
In a long-term open-label study of up to 6 months duration in 162 adolescents wearing DAYTRANA 9 hours daily, the most common (≥ 10%) adverse reactions were decreased appetite and headache. A total of 9 subjects (5.5%) were withdrawn from the study due to adverse events and 3 additional subjects (1.9%) discontinued treatment as the result of an application site reaction. Other adverse reactions leading to discontinuation that occurred with a frequency of greater than 1% included affect lability and irritability (2 subjects each, 1.2%).
6.2 Postmarketing Experience
In addition, the following adverse reactions have been identified during the post-approval use of DAYTRANA. Because these reactions are reported voluntarily from a population of uncertain size, it is not possible to reliably estimate their frequency or establish a causal relationship to DAYTRANA exposure.
**Cardiac Disorders:**palpitations.
**Eye Disorders:**visual disturbances, blurred vision, increased intraocular pressure, mydriasis, and accommodation disorder.
General Disorders and Administration Site Disorders: fatigue, application site reactions such as bleeding, bruising, burn, burning, dermatitis, discharge, discoloration, discomfort, dryness, eczema, edema, erosion, erythema, excoriation, exfoliation, fissure, hyperpigmentation, hypopigmentation, induration, infection, inflammation, irritation, pain, papules, paresthesia, pruritus, rash, scab, swelling, ulcer, urticaria, vesicles, and warmth.
Immune System Disorders: hypersensitivity reactions including generalized erythematous and urticarial rashes, allergic contact dermatitis, angioedema, and anaphylaxis.
Investigations: blood pressure increased.
Nervous System Disorders: convulsion, dyskinesia, lethargy, somnolence, serotonin syndrome in combination with serotonergic drugs, and extrapyramidal disorder, motor, and verbal tics.
Psychiatric Disorders: depression, hallucination, nervousness, and libido changes.
Skin and Subcutaneous Tissue Disorders: alopecia.
Adverse Reactions with Oral Methylphenidate Products
Nervousness and insomnia are the most common adverse reactions reported with other methylphenidate products. In children, loss of appetite, abdominal pain, weight loss during prolonged therapy, insomnia, and tachycardia may occur more frequently; however, any of the other adverse reactions listed below may also occur.
Other reactions include:
Cardiac Disorders: angina, arrhythmia, and pulse increased or decreased.
Immune System Disorders: hypersensitivity reactions including skin rash, urticaria, fever, arthralgia, exfoliative dermatitis, erythema multiforme with histopathological findings of necrotizing vasculitis, and thrombocytopenic purpura.
Metabolism and Nutrition Disorders: anorexia and weight loss during prolonged therapy.
Nervous System Disorders: drowsiness, rare reports of Tourette's syndrome and toxic psychosis.
Very rare reports of neuroleptic malignant syndrome (NMS) have been received, and, in most of these, patients were concurrently receiving therapies associated with NMS. In a single report, a ten-year-old boy who had been taking methylphenidate for approximately 18 months experienced an NMS-like event within 45 minutes of ingesting his first dose of venlafaxine. It is uncertain whether this case represented a drug-drug interaction, a response to either drug alone, or some other cause.
Vascular Disorders: blood pressure increased or decreased and cerebral arteritis and/or occlusion.
Although a definite causal relationship has not been established, the following have been reported in patients taking methylphenidate:
Blood and Lymphatic System Disorders: leukopenia and/or anemia.
Hepatobiliary Disorders: abnormal liver function, ranging from transaminase elevation to severe hepatic injury.
Psychiatric Disorders: transient depressed mood.
Skin and Subcutaneous Tissue Disorders: scalp hair loss.
Musculoskeletal and Connective Tissue Disorders: rhabdomyolysis.
- Pediatric patients (ages 6 to 12 years): The most commonly (≥5% and twice the rate of placebo) reported adverse reactions in pediatric patients ages 6 to 12 years included appetite decreased, insomnia, nausea, vomiting, weight decreased, tic, affect lability, and anorexia. (6.1)
- Pediatric patients (ages 13 to 17 years): The most commonly (≥5% and twice the rate of placebo) reported adverse reactions in pediatric patients ages 13 to 17 years included appetite decreased, nausea, insomnia, weight decreased, dizziness, abdominal pain, and anorexia. The majority of subjects in these trials had erythema at the application site. (6.1)
To report SUSPECTED ADVERSE REACTIONS, contact Noven Therapeutics, LLC at 1-877-567-7857 or FDA at 1-800-FDA-1088 orwww.fda.gov/medwatch.
DRUG ABUSE AND DEPENDENCE SECTION
9 DRUG ABUSE AND DEPENDENCE
9.1 Controlled Substance
DAYTRANA contains methylphenidate, a Schedule II controlled substance.
9.2 Abuse
DAYTRANA has a high potential for abuse and misuse which can lead to the development of a substance use disorder, including addiction [see Warnings and Precautions (5.1)]. DAYTRANA can be diverted for non-medical use into illicit channels or distribution.
Abuse is the intentional non-therapeutic use of a drug, even once, to achieve a desired psychological or physiological effect. Misuse is the intentional use, for therapeutic purposes, of a drug by an individual in a way other than prescribed by a health care provider or for whom it was not prescribed. Drug addiction is a cluster of behavioral, cognitive, and physiological phenomena that may include a strong desire to take the drug, difficulties in controlling drug use (e.g., continuing drug use despite harmful consequences, giving a higher priority to drug use than other activities and obligations), and possible tolerance or physical dependence.
Misuse and abuse of methylphenidate may cause increased heart rate, respiratory rate, or blood pressure; sweating; dilated pupils; hyperactivity; restlessness; insomnia; decreased appetite; loss of coordination; tremors; flushed skin; vomiting; and/or abdominal pain. Anxiety, psychosis, hostility, aggression, and suicidal or homicidal ideation have also been observed with CNS stimulants abuse and/or misuse. Misuse and abuse of CNS stimulants, including DAYTRANA, can result in overdose and death [see Overdosage (10)], and this risk is increased with higher doses or unapproved methods of administration, such as snorting or injection.
9.3 Dependence
Physical Dependence
DAYTRANA may produce physical dependence. Physical dependence is a state that develops as a result of physiological adaptation in response to repeated drug use, manifested by withdrawal signs and symptoms after abrupt discontinuation or a significant dose reduction of a drug.
Withdrawal signs and symptoms after abrupt discontinuation or dose reduction following prolonged use of CNS stimulants including DAYTRANA include dysphoric mood; depression; fatigue; vivid, unpleasant dreams; insomnia or hypersomnia; increased appetite; and psychomotor retardation or agitation.
Tolerance
DAYTRANA may produce tolerance. Tolerance is a physiological state characterized by a reduced response to a drug after repeated administration (i.e., a higher dose of a drug is required to produce the same effect that was once obtained at a lower dose).
DESCRIPTION SECTION
11 DESCRIPTION
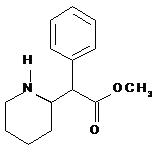
DAYTRANA is an adhesive-based matrix transdermal system containing methylphenidate that is applied to intact skin. The chemical name for methylphenidate is α-phenyl-2-piperidineacetic acid methyl ester. It is a white to off-white powder and is soluble in alcohol, ethyl acetate, and ether. Methylphenidate is practically insoluble in water and petrol ether. Its molecular weight is 233.31. Its empirical formula is C14H19NO2. The structural formula of methylphenidate is:
Transdermal System Components
DAYTRANA contains methylphenidate in a multipolymeric adhesive. The methylphenidate is dispersed in acrylic adhesive that is dispersed in a silicone adhesive. The composition per unit area of all dosage strengths is identical, and the total dose delivered is dependent on the transdermal system size and wear time.
DAYTRANA consists of three layers, as seen in the figure below (cross-section of the transdermal system).
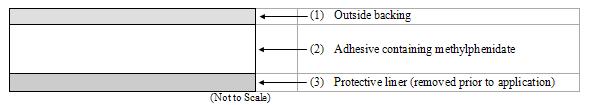
Proceeding from the outer surface toward the surface adhering to the skin, the layers are (1) a polyester/ethylene vinyl acetate laminate film backing, (2) a proprietary adhesive formulation incorporating Noven Pharmaceuticals, Inc.'s DOT Matrix™ transdermal technology consisting of an acrylic adhesive, a silicone adhesive, and methylphenidate, and (3) a fluoropolymer-coated polyester protective liner, which is attached to the adhesive surface and must be removed before the transdermal system can be used.
The active component of the transdermal system is methylphenidate. The remaining components are pharmacologically inactive.
NONCLINICAL TOXICOLOGY SECTION
13 NONCLINICAL TOXICOLOGY
13.1 Carcinogenesis/Mutagenesis and Impairment of Fertility
Carcinogenesis
Carcinogenicity studies of transdermal methylphenidate have not been performed. In a lifetime carcinogenicity study of oral methylphenidate carried out in B6C3F1 mice, methylphenidate caused an increase in hepatocellular adenomas and, in males only, an increase in hepatoblastomas, at a daily dose of approximately 60 mg/kg/day. Hepatoblastoma is a relatively rare rodent malignant tumor type. There was no increase in total malignant hepatic tumors. The mouse strain used is sensitive to the development of hepatic tumors and the significance of these results to humans is unknown.
Orally administered methylphenidate did not cause any increases in tumors in a lifetime carcinogenicity study carried out in F344 rats; the highest dose used was approximately 45 mg/kg/day.
In a 24-week oral carcinogenicity study in the transgenic mouse strain p53+/-, which is sensitive to genotoxic carcinogens, there was no evidence of carcinogenicity. In this study, male and female mice were fed diets containing the same concentration of methylphenidate as in the lifetime carcinogenicity study; the high-dose groups were exposed to 60 to 74 mg/kg/day of methylphenidate.
Mutagenesis
Methylphenidate was not mutagenic in the in vitro Ames reverse mutation assay or in the in vitro mouse lymphoma cell forward mutation assay. Sister chromatid exchanges and chromosome aberrations were increased, indicative of a weak clastogenic response, in an in vitro assay in cultured Chinese hamster ovary cells. Methylphenidate was negative in vivo in males and females in the mouse bone marrow micronucleus assay.
Impairment of Fertility
Methylphenidate did not impair fertility in male or female mice that were fed diets containing the drug in an 18-week continuous breeding study. The study was conducted at doses up to 160 mg/kg/day.
HOW SUPPLIED SECTION
16 HOW SUPPLIED/STORAGE AND HANDLING
DAYTRANA is supplied in a sealed tray containing 30 individually pouched transdermal systems. See the chart below for information regarding available strengths.
|
*Nominal in vivo delivery rate per hour in children and adolescents when applied to the hip, based on a 9-hour wear period. **Methylphenidate content in each transdermal system. | |||||
|
Nominal Dose |
Dosage |
Transdermal System |
Methylphenidate |
Transdermal Systems |
NDC Number |
|
10 |
1.1 |
12.5 |
27.5 |
30 |
68968-5552-3 |
|
15 |
1.6 |
18.75 |
41.3 |
30 |
68968-5553-3 |
|
20 |
2.2 |
25 |
55 |
30 |
68968-5554-3 |
|
30 |
3.3 |
37.5 |
82.5 |
30 |
68968-5555-3 |
Store at 25° C (77° F); excursions permitted to 15-30° C (59-86° F) [see USP Controlled Room Temperature]. Do not store transdermal systems unpouched. Do not store transdermal systems in refrigerators or freezers.
Once the sealed tray is opened, use contents within 2 months. Apply the transdermal system immediately upon removal from the individual protective pouch. For transdermal use only.
See the Patient Counseling Information (17) for specific disposal instructions for unused or expired DAYTRANA.
SPL MEDGUIDE SECTION
|
This Medication Guide has been approved by the U.S. Food and Drug Administration. |
Revised 10/2023 | |
|
MEDICATION GUIDE DAYTRANA**®**** (day-TRON-ah)** | ||
|
Important: DAYTRANA is for use on the skin only. | ||
|
What is the most important information I should know about DAYTRANA? DAYTRANA may cause serious side effects, including: ***Abuse, misuse, and addiction.**DAYTRANA has a high chance for abuse and misuse and may lead to substance use problems, including addiction. Misuse and abuse of DAYTRANA, other methylphenidate containing medicines, and amphetamine containing medicines, can lead to overdose and death. The risk of overdose and death is increased with higher doses of DAYTRANA or when it is used in ways that are not approved, such as snorting or injection.
* Your healthcare provider should check your child's risk for abuse, misuse, and addiction before starting treatment with DAYTRANA and will monitor your child during treatment.
* DAYTRANA may lead to physical dependence after prolonged use, even if taken as directed by your healthcare provider.
* Do not give DAYTRANA to anyone else. See "What is DAYTRANA?" for more information.
* Keep DAYTRANA in a safe place and properly dispose of any unused medicine. See "How should I store DAYTRANA?" for more information.
* Tell your healthcare provider if your child has ever abused or been dependent on alcohol, prescription medicines, or street drugs.
*Risks for people with serious heart disease. Sudden death has happened in people who have heart defects or other serious heart disease. *Increased blood pressure and heart rate. *Mental (psychiatric) problems, including:
* new or worse behavior or thought problems
* new or worse bipolar illness
* new psychotic symptoms (such as hearing voices, or seeing or believing things that are not real) or manic symptoms
Tell your child's healthcare provider about any mental problems your child has
or about a family history of suicide, bipolar illness, or depression. | ||
|
What Is DAYTRANA? DAYTRANA is a central nervous system (CNS) stimulant prescription medication used for the treatment of Attention Deficit Hyperactivity Disorder (ADHD) in children 6 to 17 years of age. DAYTRANA may help increase attention and decrease impulsiveness and hyperactivity in children with ADHD. It is not known if DAYTRANA is safe and effective in children younger than 6 years. DAYTRANA is a federally controlled substance (CII) because it contains methylphenidate that can be a target for people who abuse prescription medicines or street drugs. Keep DAYTRANA in a safe place to protect it from theft. Never give your DAYTRANA to anyone else because it may cause death or harm them. Selling or giving away DAYTRANA may harm others and is against the law. | ||
|
Do not use DAYTRANA if your child:
| ||
|
Before using DAYTRANA, tell your child's healthcare provider about all of your child's medical conditions, including if your child:
**Tell your child's healthcare provider about all of the medicines your child takes,**including prescription and over-the-counter medicines, vitamins, and herbal supplements. DAYTRANA and some medicines may interact with each other and cause serious side effects. Sometimes the doses of other medicines will need to be changed during treatment with DAYTRANA. Your child's healthcare provider will decide if DAYTRANA can be taken with other medicines. Especially tell your child's healthcare provider if your child takes:
Know the medicines that your child takes. Keep a list of your child's medicines with you to show your child's healthcare provider and pharmacist when your child gets a new medicine.Do not start any new medicine while using DAYTRANA without first talking to your child's healthcare provider. | ||
|
How should DAYTRANA be used?
If your child uses too much DAYTRANA transdermal systems call your healthcare provider or Poison Help line at 1-800-222-1222 or go to the nearest hospital emergency room right away. | ||
|
What should your child avoid while using DAYTRANA?
| ||
|
What are the possible side effects of DAYTRANA? DAYTRANA may cause serious side effects, including:
*Slowing of growth (height and weight) in children. Your child should have their height and weight checked often during treatment with DAYTRANA. Your healthcare provider may stop your child’s DAYTRANA treatment if they are not growing or gaining weight as expected. *Eyes problems (increased pressure in the eye and glaucoma). Call your healthcare provider right away if you or your child develop changes in your vision or eye pain, swelling, or redness. *New or worsening tics or worsening Tourette's syndrome. Tell your healthcare provider if you or your child get any new or worsening tics or worsening Tourettes syndrome during treatment with DAYTRANA. *Loss of skin color. DAYTRANA may cause a persistent loss of skin-color where the patch is applied or around the patch application site. Loss of skin-color, in some cases, has been reported at locations on the skin far from any application site. The loss of skin-color may be permanent even after removing the patch or DAYTRANA is stopped. Call your healthcare provider right away if your child has changes in skin-color. DAYTRANA treatment may be stopped if your child has changes in skin color. *Allergic skin rash (contact sensitization). Stop using DAYTRANA and tell your child's healthcare provider right away if your child develops swelling or blisters at or around the application site. Your child may have a skin allergy to DAYTRANA. People who have skin allergies to DAYTRANA may develop an allergy to all medicines that contain methylphenidate, even methylphenidate medicines taken by mouth. | ||
|
The most common side effects of DAYTRANA in children 6 to 12 years old include: | ||
|
|
|
|
The most common side effects of DAYTRANA in children 13 to 17 years old include: | ||
|
|
|
|
DAYTRANA may also cause skin problems where it is applied (redness, small bumps, itching) Your child's doctor may do certain blood tests while your child uses DAYTRANA. These are not all the possible side effects of DAYTRANA. Call your doctor for medical advice about side effects. You may report side effects to FDA at 1-800-FDA-1088. | ||
|
How should I store DAYTRANA?
Keep DAYTRANA and all medicines out of the reach of children. | ||
|
General information about the safe and effective use of DAYTRANA. Medicines are sometimes prescribed for purposes other than those listed in a Medication Guide. Do not use DAYTRANA for a condition for which it was not prescribed. Do not give DAYTRANA to other people, even if they have the same symptoms. It may harm them and it is against the law. You can ask your pharmacist or healthcare provider for information about DAYTRANA that is written for healthcare professionals. | ||
|
What are the ingredients in DAYTRANA? Manufactured by: Noven Pharmaceuticals, Inc., Miami, FL 33186 For more information, go to www.daytrana.com, or call 1-877-567-7857. |