
Divalproex sodium
These highlights do not include all the information needed to use DIVALPROEX SODIUM EXTENDED-RELEASE TABLETS safely and effectively. See full prescribing information for DIVALPROEX SODIUM EXTENDED-RELEASE TABLETS. DIVALPROEX SODIUM extended-release tablets, for oral use Initial U.S. Approval: 2000
94eac383-a89f-4128-b81c-076640b8edcf
HUMAN PRESCRIPTION DRUG LABEL
Aug 24, 2023
A-S Medication Solutions
DUNS: 830016429
Products 1
Detailed information about drug products covered under this FDA approval, including NDC codes, dosage forms, ingredients, and administration routes.
Divalproex sodium
Product Details
FDA regulatory identification and product classification information
FDA Identifiers
Product Classification
Product Specifications
INGREDIENTS (19)
Drug Labeling Information
Drug Interactions Section
7 DRUG INTERACTIONS
7.1 Effects of Co-Administered Drugs on Valproate Clearance
Drugs that affect the level of expression of hepatic enzymes, particularly those that elevate levels of glucuronosyltransferases (such as ritonavir), may increase the clearance of valproate. For example, phenytoin, carbamazepine, and phenobarbital (or primidone) can double the clearance of valproate. Thus, patients on monotherapy will generally have longer half-lives and higher concentrations than patients receiving polytherapy with antiepilepsy drugs.
In contrast, drugs that are inhibitors of cytochrome P450 isozymes, e.g., antidepressants, may be expected to have little effect on valproate clearance because cytochrome P450 microsomal mediated oxidation is a relatively minor secondary metabolic pathway compared to glucuronidation and beta-oxidation.
Because of these changes in valproate clearance, monitoring of valproate and concomitant drug concentrations should be increased whenever enzyme inducing drugs are introduced or withdrawn.
The following list provides information about the potential for an influence of several commonly prescribed medications on valproate pharmacokinetics. The list is not exhaustive nor could it be, since new interactions are continuously being reported.
Drugs for which a potentially important interaction has been observed
Aspirin
A study involving the co-administration of aspirin at antipyretic doses (11 to 16 mg/kg) with valproate to pediatric patients (n=6) revealed a decrease in protein binding and an inhibition of metabolism of valproate. Valproate free fraction was increased 4-fold in the presence of aspirin compared to valproate alone. The β-oxidation pathway consisting of 2-E-valproic acid, 3-OH-valproic acid, and 3-keto valproic acid was decreased from 25% of total metabolites excreted on valproate alone to 8.3% in the presence of aspirin. Whether or not the interaction observed in this study applies to adults is unknown, but caution should be observed if valproate and aspirin are to be co-administered.
Carbapenem Antibiotics
A clinically significant reduction in serum valproic acid concentration has been reported in patients receiving carbapenem antibiotics (for example, ertapenem, imipenem, meropenem; this is not a complete list) and may result in loss of seizure control. The mechanism of this interaction is not well understood. Serum valproic acid concentrations should be monitored frequently after initiating carbapenem therapy. Alternative antibacterial or anticonvulsant therapy should be considered if serum valproic acid concentrations drop significantly or seizure control deteriorates [see Warnings and Precautions (5.13)].
Estrogen-Containing Hormonal Contraceptives
Estrogen-containing hormonal contraceptives may increase the clearance of valproate, which may result in decreased concentration of valproate and potentially increased seizure frequency. Prescribers should monitor serum valproate concentrations and clinical response when adding or discontinuing estrogen containing products.
Felbamate
A study involving the co-administration of 1,200 mg/day of felbamate with valproate to patients with epilepsy (n=10) revealed an increase in mean valproate peak concentration by 35% (from 86 to 115 mcg/mL) compared to valproate alone. Increasing the felbamate dose to 2,400 mg/day increased the mean valproate peak concentration to 133 mcg/mL (another 16% increase). A decrease in valproate dosage may be necessary when felbamate therapy is initiated.
Methotrexate
Methotrexate may decrease serum valproate levels and potentially result in increased frequency of seizures or bipolar symptoms. Prescribers should monitor serum valproate concentrations and clinical response when adding or discontinuing methotrexate and adjust valproate dosage, if necessary.
Rifampin
A study involving the administration of a single dose of valproate (7 mg/kg) 36 hours after 5 nights of daily dosing with rifampin (600 mg) revealed a 40% increase in the oral clearance of valproate. Valproate dosage adjustment may be necessary when it is co-administered with rifampin.
7.2 Effects of Valproate on Other Drugs
Valproate has been found to be a weak inhibitor of some P450 isozymes, epoxide hydrase, and glucuronosyltransferases.
The following list provides information about the potential for an influence of valproate co-administration on the pharmacokinetics or pharmacodynamics of several commonly prescribed medications. The list is not exhaustive, since new interactions are continuously being reported.
Drugs for which a potentially important valproate interaction has been observed
Amitriptyline/Nortriptyline
Administration of a single oral 50 mg dose of amitriptyline to 15 normal volunteers (10 males and 5 females) who received valproate (500 mg BID) resulted in a 21% decrease in plasma clearance of amitriptyline and a 34% decrease in the net clearance of nortriptyline. Rare postmarketing reports of concurrent use of valproate and amitriptyline resulting in an increased amitriptyline level have been received. Concurrent use of valproate and amitriptyline has rarely been associated with toxicity. Monitoring of amitriptyline levels should be considered for patients taking valproate concomitantly with amitriptyline. Consideration should be given to lowering the dose of amitriptyline/nortriptyline in the presence of valproate.
Carbamazepine/carbamazepine-10,11-Epoxide
Serum levels of carbamazepine (CBZ) decreased 17% while that of carbamazepine-10,11-epoxide (CBZ-E) increased by 45% upon co-administration of valproate and CBZ to epileptic patients.
Clonazepam
The concomitant use of valproate and clonazepam may induce absence status in patients with a history of absence type seizures.
Diazepam
Valproate displaces diazepam from its plasma albumin binding sites and inhibits its metabolism. Co-administration of valproate (1,500 mg daily) increased the free fraction of diazepam (10 mg) by 90% in healthy volunteers (n=6). Plasma clearance and volume of distribution for free diazepam were reduced by 25% and 20%, respectively, in the presence of valproate. The elimination half-life of diazepam remained unchanged upon addition of valproate.
Ethosuximide
Valproate inhibits the metabolism of ethosuximide. Administration of a single ethosuximide dose of 500 mg with valproate (800 to 1,600 mg/day) to healthy volunteers (n=6) was accompanied by a 25% increase in elimination half-life of ethosuximide and a 15% decrease in its total clearance as compared to ethosuximide alone. Patients receiving valproate and ethosuximide, especially along with other anticonvulsants, should be monitored for alterations in serum concentrations of both drugs.
Lamotrigine
In a steady-state study involving 10 healthy volunteers, the elimination half- life of lamotrigine increased from 26 to 70 hours with valproate co- administration (a 165% increase). The dose of lamotrigine should be reduced when co-administered with valproate. Serious skin reactions (such as Stevens- Johnson syndrome and toxic epidermal necrolysis) have been reported with concomitant lamotrigine and valproate administration. See lamotrigine package insert for details on lamotrigine dosing with concomitant valproate administration.
Phenobarbital
Valproate was found to inhibit the metabolism of phenobarbital. Co- administration of valproate (250 mg BID for 14 days) with phenobarbital to normal subjects (n=6) resulted in a 50% increase in half-life and a 30% decrease in plasma clearance of phenobarbital (60 mg single-dose). The fraction of phenobarbital dose excreted unchanged increased by 50% in presence of valproate.
There is evidence for severe CNS depression, with or without significant elevations of barbiturate or valproate serum concentrations. All patients receiving concomitant barbiturate therapy should be closely monitored for neurological toxicity. Serum barbiturate concentrations should be obtained, if possible, and the barbiturate dosage decreased, if appropriate.
Primidone, which is metabolized to a barbiturate, may be involved in a similar interaction with valproate.
Phenytoin
Valproate displaces phenytoin from its plasma albumin binding sites and inhibits its hepatic metabolism. Co-administration of valproate (400 mg TID) with phenytoin (250 mg) in normal volunteers (n=7) was associated with a 60% increase in the free fraction of phenytoin. Total plasma clearance and apparent volume of distribution of phenytoin increased 30% in the presence of valproate. Both the clearance and apparent volume of distribution of free phenytoin were reduced by 25%.
In patients with epilepsy, there have been reports of breakthrough seizures occurring with the combination of valproate and phenytoin. The dosage of phenytoin should be adjusted as required by the clinical situation.
Propofol
The concomitant use of valproate and propofol may lead to increased blood levels of propofol. Reduce the dose of propofol when co-administering with valproate. Monitor patients closely for signs of increased sedation or cardiorespiratory depression.
Rufinamide
Based on a population pharmacokinetic analysis, rufinamide clearance was decreased by valproate. Rufinamide concentrations were increased by <16% to 70%, dependent on concentration of valproate (with the larger increases being seen in pediatric patients at high doses or concentrations of valproate). Patients stabilized on rufinamide before being prescribed valproate should begin valproate therapy at a low dose, and titrate to a clinically effective dose [see Dosage and Administration (2.6)]. Similarly, patients on valproate should begin at a rufinamide dose lower than 10 mg/kg per day (pediatric patients) or 400 mg per day (adults).
Tolbutamide
From in vitro experiments, the unbound fraction of tolbutamide was increased from 20% to 50% when added to plasma samples taken from patients treated with valproate. The clinical relevance of this displacement is unknown.
Warfarin
In an in vitro study, valproate increased the unbound fraction of warfarin by up to 32.6%. The therapeutic relevance of this is unknown; however, coagulation tests should be monitored if valproate therapy is instituted in patients taking anticoagulants.
Zidovudine
In six patients who were seropositive for HIV, the clearance of zidovudine (100 mg q8h) was decreased by 38% after administration of valproate (250 or 500 mg q8h); the half-life of zidovudine was unaffected.
7.3 Topiramate
Concomitant administration of valproate and topiramate has been associated with hyperammonemia with and without encephalopathy [see Contraindications (4) and Warnings and Precautions (5.6, 5.9, 5.10)]. Concomitant administration of topiramate with valproate has also been associated with hypothermia in patients who have tolerated either drug alone. It may be prudent to examine blood ammonia levels in patients in whom the onset of hypothermia has been reported [see Warnings and Precautions (5.9, 5.11)].
7.4 Cannabidiol
Concomitant administration of valproate and cannabidiol has been associated with an increased risk of ALT and/or AST elevation. This has been manageable by dose reduction or, in more severe cases, by discontinuation of one or both drugs. Liver function, including serum transaminase and total bilirubin levels, should be monitored during concomitant treatment [see Warnings and Precautions (5.1)].
- Hepatic enzyme-inducing drugs (e.g., phenytoin, carbamazepine, phenobarbital, primidone, rifampin) can increase valproate clearance, while enzyme inhibitors (e.g., felbamate) can decrease valproate clearance. Therefore increased monitoring of valproate and concomitant drug concentrations and dosage adjustment are indicated whenever enzyme-inducing or inhibiting drugs are introduced or withdrawn (7.1)
- Aspirin, carbapenem antibiotics, estrogen-containing hormonal contraceptives, methotrexate: Monitoring of valproate concentrations is recommended (7.1)
- Co-administration of valproate can affect the pharmacokinetics of other drugs (e.g., diazepam, ethosuximide, lamotrigine, phenytoin) by inhibiting their metabolism or protein binding displacement (7.2)
- Patients stabilized on rufinamide should begin valproate therapy at a low dose, and titrate to clinically effective dose (7.2)
- Dosage adjustment of amitriptyline/nortriptyline, propofol, warfarin, and zidovudine may be necessary if used concomitantly with divalproex sodium extended-release tablets (7.2)
- Topiramate: Hyperammonemia and encephalopathy (5.10, 7.3)
- Cannabidiol: ALT and/or AST elevation (7.4)
Clinical Pharmacology Section
12 CLINICAL PHARMACOLOGY
12.1 Mechanism of Action
Divalproex sodium dissociates to the valproate ion in the gastrointestinal tract. The mechanisms by which valproate exerts its therapeutic effects have not been established. It has been suggested that its activity in epilepsy is related to increased brain concentrations of gamma-aminobutyric acid (GABA).
12.2 Pharmacodynamics
The relationship between plasma concentration and clinical response is not well documented. One contributing factor is the nonlinear, concentration dependent protein binding of valproate which affects the clearance of the drug. Thus, monitoring of total serum valproate may not provide a reliable index of the bioactive valproate species.
For example, because the plasma protein binding of valproate is concentration dependent, the free fraction increases from approximately 10% at 40 mcg/mL to 18.5% at 130 mcg/mL. Higher than expected free fractions occur in the elderly, in hyperlipidemic patients, and in patients with hepatic and renal diseases.
Epilepsy
The therapeutic range in epilepsy is commonly considered to be 50 to 100 mcg/mL of total valproate, although some patients may be controlled with lower or higher plasma concentrations.
Mania
In placebo-controlled clinical trials of acute mania, patients were dosed to clinical response with trough plasma concentrations between 85 and 125 mcg/mL [see Dosage and Administration (2.1)].
12.3 Pharmacokinetics
Absorption/Bioavailability
The absolute bioavailability of divalproex sodium extended-release tablets administered as a single dose after a meal was approximately 90% relative to intravenous infusion.
When given in equal total daily doses, the bioavailability of divalproex sodium extended-release tablets is less than that of divalproex sodium delayed-release tablets. In five multiple-dose studies in healthy subjects (N=82) and in subjects with epilepsy (N=86), when administered under fasting and nonfasting conditions, divalproex sodium extended-release tablets given once daily produced an average bioavailability of 89% relative to an equal total daily dose of divalproex sodium delayed-release tablets given BID, TID, or QID. The median time to maximum plasma valproate concentrations (Cmax) after divalproex sodium extended-release tablets administration ranged from 4 to 17 hours. After multiple once-daily dosing of divalproex sodium extended- release tablets, the peak-to-trough fluctuation in plasma valproate concentrations was 10 to 20% lower than that of regular divalproex sodium delayed-release tablets given BID, TID, or QID.
** Conversion from Divalproex Sodium Delayed-Release Tablets to Divalproex Sodium Extended-Release Tablets**
When divalproex sodium extended-release tablets are given in doses 8 to 20% higher than the total daily dose of divalproex sodium delayed-release tablets, the two formulations are bioequivalent. In two randomized, crossover studies, multiple daily doses of divalproex sodium delayed-release tablets were compared to 8 to 20% higher once-daily doses of divalproex sodium extended- release tablets. In these two studies, divalproex sodium extended-release tablets and divalproex sodium delayed-release tablets regimens were equivalent with respect to area under the curve (AUC; a measure of the extent of bioavailability). Additionally, valproate Cmax was lower, and Cmin was either higher or not different, for divalproex sodium extended-release tablets relative to divalproex sodium delayed-release tablets regimens (see Table 8).
Table 8. Bioavailability of Divalproex Sodium Extended-Release Tablets Relative to Divalproex Sodium Delayed-Release Tablets When Divalproex Sodium Extended-Release Tablets Dose is 8 to 20% Higher|
Study Population |
Regimens |
Relative Bioavailability | ||
|---|---|---|---|---|
|
Divalproex Sodium |
AUC24 |
Cmax |
Cmin | |
|
Healthy Volunteers (N=35) |
1,000 & 1,500 mg |
1.059 |
0.882 |
1.173 |
|
Patients with epilepsy on concomitant enzyme- |
1,000 to 5,000 mg |
1.008 |
0.899 |
1.022 |
Concomitant antiepilepsy drugs (topiramate, phenobarbital, carbamazepine, phenytoin, and lamotrigine were evaluated) that induce the cytochrome P450 isozyme system did not significantly alter valproate bioavailability when converting between divalproex sodium delayed-release tablets and divalproex sodium extended-release tablets.
Distribution
Protein Binding
The plasma protein binding of valproate is concentration dependent and the free fraction increases from approximately 10% at 40 mcg/mL to 18.5% at 130 mcg/mL. Protein binding of valproate is reduced in the elderly, in patients with chronic hepatic diseases, in patients with renal impairment, and in the presence of other drugs (e.g., aspirin). Conversely, valproate may displace certain protein-bound drugs (e.g., phenytoin, carbamazepine, warfarin, and tolbutamide) [see Drug Interactions (7.2) for more detailed information on the pharmacokinetic interactions of valproate with other drugs].
CNS Distribution
Valproate concentrations in cerebrospinal fluid (CSF) approximate unbound concentrations in plasma (about 10% of total concentration).
Metabolism
Valproate is metabolized almost entirely by the liver. In adult patients on
monotherapy, 30 to 50% of an administered dose appears in urine as a
glucuronide conjugate. Mitochondrial β-oxidation is the other major metabolic
pathway, typically accounting for over 40% of the dose. Usually, less than 15
to 20% of the dose is eliminated by other oxidative mechanisms. Less than 3%
of an administered dose is excreted unchanged in urine.
The relationship between dose and total valproate concentration is nonlinear; concentration does not increase proportionally with the dose, but rather, increases to a lesser extent due to saturable plasma protein binding. The kinetics of unbound drug are linear.
Elimination
Mean plasma clearance and volume of distribution for total valproate are 0.56
L/hr/1.73 m2 and 11 L/1.73 m2, respectively. Mean plasma clearance and volume
of distribution for free valproate are 4.6 L/hr/1.73 m2 and 92 L/1.73 m2. Mean
terminal half-life for valproate monotherapy ranged from 9 to 16 hours
following oral dosing regimens of 250 to 1,000 mg.
The estimates cited apply primarily to patients who are not taking drugs that affect hepatic metabolizing enzyme systems. For example, patients taking enzyme-inducing antiepileptic drugs (carbamazepine, phenytoin, and phenobarbital) will clear valproate more rapidly. Because of these changes in valproate clearance, monitoring of antiepileptic concentrations should be intensified whenever concomitant antiepileptics are introduced or withdrawn.
Specific Populations
Effect of Age
Pediatric
The valproate pharmacokinetic profile following administration of divalproex sodium extended-release tablets was characterized in a multiple-dose, non- fasting, open label, multi-center study in children and adolescents. Divalproex sodium extended-release tablets once daily doses ranged from 250 to 1,750 mg. Once daily administration of divalproex sodium extended-release tablets in pediatric patients (10 to 17 years) produced plasma VPA concentration-time profiles similar to those that have been observed in adults.
Elderly
The capacity of elderly patients (age range: 68 to 89 years) to eliminate valproate has been shown to be reduced compared to younger adults (age range: 22 to 26 years). Intrinsic clearance is reduced by 39%; the free fraction is increased by 44%. Accordingly, the initial dosage should be reduced in the elderly [see Dosage and Administration (2.4)].
Effect of Sex
There are no differences in the body surface area adjusted unbound clearance
between males and females (4.8±0.17 and 4.7±0.07 L/hr per 1.73 m2,
respectively).
Effect of Race
The effects of race on the kinetics of valproate have not been studied.
Effect of Disease
Liver Disease
Liver disease impairs the capacity to eliminate valproate. In one study, the clearance of free valproate was decreased by 50% in 7 patients with cirrhosis and by 16% in 4 patients with acute hepatitis, compared with 6 healthy subjects. In that study, the half-life of valproate was increased from 12 to 18 hours. Liver disease is also associated with decreased albumin concentrations and larger unbound fractions (2 to 2.6 fold increase) of valproate. Accordingly, monitoring of total concentrations may be misleading since free concentrations may be substantially elevated in patients with hepatic disease whereas total concentrations may appear to be normal [see Boxed Warning, Contraindications (4), and Warnings and Precautions (5.1)].
Renal Disease
A slight reduction (27%) in the unbound clearance of valproate has been reported in patients with renal failure (creatinine clearance < 10 mL/minute); however, hemodialysis typically reduces valproate concentrations by about 20%. Therefore, no dosage adjustment appears to be necessary in patients with renal failure. Protein binding in these patients is substantially reduced; thus, monitoring total concentrations may be misleading.
Drug Interaction Studies with No Interaction or Likely Clinically Unimportant Interaction
Antacids
A study involving the co-administration of valproate 500 mg with commonly administered antacids (Maalox, Trisogel, and Titralac - 160 mEq doses) did not reveal any effect on the extent of absorption of valproate.
Chlorpromazine
A study involving the administration of 100 to 300 mg/day of chlorpromazine to schizophrenic patients already receiving valproate (200 mg BID) revealed a 15% increase in trough plasma levels of valproate.
Haloperidol
A study involving the administration of 6 to 10 mg/day of haloperidol to schizophrenic patients already receiving valproate (200 mg BID) revealed no significant changes in valproate trough plasma levels.
Cimetidine and Ranitidine
Cimetidine and ranitidine do not affect the clearance of valproate.
Acetaminophen
Valproate had no effect on any of the pharmacokinetic parameters of acetaminophen when it was concurrently administered to three epileptic patients.
Clozapine
In psychotic patients (n=11), no interaction was observed when valproate was co-administered with clozapine.
Lithium
Co-administration of valproate (500 mg BID) and lithium carbonate (300 mg TID) to normal male volunteers (n=16) had no effect on the steady-state kinetics of lithium.
Lorazepam
Concomitant administration of valproate (500 mg BID) and lorazepam (1 mg BID) in normal male volunteers (n=9) was accompanied by a 17% decrease in the plasma clearance of lorazepam.
Olanzapine
No dose adjustment for olanzapine is necessary when olanzapine is administered concomitantly with valproate. Co-administration of valproate (500 mg BID) and olanzapine (5 mg) to healthy adults (n=10) caused 15% reduction in Cmax and 35% reduction in AUC of olanzapine.
Oral Contraceptive Steroids
Administration of a single-dose of ethinyloestradiol (50 mcg)/levonorgestrel (250 mcg) to 6 women on valproate (200 mg BID) therapy for 2 months did not reveal any pharmacokinetic interaction.
Nonclinical Toxicology Section
13 NONCLINICAL TOXICOLOGY
13.1 Carcinogenesis, Mutagenesis, Impairment of Fertility
Carcinogenesis
Valproate was administered orally to rats and mice at doses of 80 and 170 mg/kg/day (less than the maximum recommended human dose on a mg/m2 basis) for two years. The primary findings were an increase in the incidence of subcutaneous fibrosarcomas in high-dose male rats receiving valproate and a dose-related trend for benign pulmonary adenomas in male mice receiving valproate.
Mutagenesis
Valproate was not mutagenic in an in vitro bacterial assay (Ames test), did not produce dominant lethal effects in mice, and did not increase chromosome aberration frequency in an in vivo cytogenetic study in rats. Increased frequencies of sister chromatid exchange (SCE) have been reported in a study of epileptic children taking valproate; this association was not observed in another study conducted in adults.
Impairment of Fertility
In chronic toxicity studies in juvenile and adult rats and dogs, administration of valproate resulted in testicular atrophy and reduced spermatogenesis at oral doses of 400 mg/kg/day or greater in rats (approximately equal to or greater than the maximum recommended human dose (MRHD) on a mg/m2 basis) and 150 mg/kg/day or greater in dogs (approximately equal to or greater than the MRHD on a mg/m2 basis). Fertility studies in rats have shown no effect on fertility at oral doses of valproate up to 350 mg/kg/day (approximately equal to the MRHD on a mg/m2 basis) for 60 days.
Clinical Studies Section
14 CLINICAL STUDIES
14.1 Mania
The effectiveness of divalproex sodium extended-release tablets for the treatment of acute mania is based in part on studies establishing the effectiveness of divalproex sodium delayed-release tablets for this indication. Divalproex sodium extended-release tablet’s effectiveness was confirmed in one randomized, double-blind, placebo-controlled, parallel group, 3-week, multicenter study. The study was designed to evaluate the safety and efficacy of divalproex sodium extended-release tablets in the treatment of bipolar I disorder, manic or mixed type, in adults. Adult male and female patients who had a current DSM-IV TR primary diagnosis of bipolar I disorder, manic or mixed type, and who were hospitalized for acute mania, were enrolled into this study. Divalproex sodium extended-release tablets were initiated at a dose of 25 mg/kg/day given once daily, increased by 500 mg/day on Day 3, then adjusted to achieve plasma valproate concentrations in the range of 85 to 125 mcg/mL. Mean daily divalproex sodium extended-release tablets dose for observed cases were 2,362 mg (range: 500 to 4,000), 2,874 mg (range: 1,500 to 4,500), 2,993 mg (range: 1,500 to 4,500), 3,181 mg (range: 1,500 to 5,000), and 3,353 mg (range: 1,500 to 5,500) at Days 1, 5, 10, 15, and 21, respectively. Mean valproate concentrations were 96.5 mcg/mL, 102.1 mcg/mL, 98.5 mcg/mL, 89.5 mcg/mL at Days 5, 10, 15 and 21, respectively. Patients were assessed on the Mania Rating Scale (MRS; score ranges from 0 to 52).
Divalproex sodium extended-release tablets were significantly more effective than placebo in reduction of the MRS total score.
14.2 Epilepsy
The efficacy of valproate in reducing the incidence of complex partial seizures (CPS) that occur in isolation or in association with other seizure types was established in two controlled trials.
In one, multi-clinic, placebo controlled study employing an add-on design (adjunctive therapy), 144 patients who continued to suffer eight or more CPS per 8 weeks during an 8 week period of monotherapy with doses of either carbamazepine or phenytoin sufficient to assure plasma concentrations within the “therapeutic range” were randomized to receive, in addition to their original antiepilepsy drug (AED), either divalproex sodium delayed-release tablets or placebo. Randomized patients were to be followed for a total of 16 weeks. The following table presents the findings.
Table 9. Adjunctive Therapy Study Median Incidence of CPS per 8 Weeks
| |||
|
** Add-on Treatment** |
Number |
Baseline |
Experimental Incidence |
|
Divalproex Sodium Delayed-Release Tablets |
75 |
16 |
8.9* |
|
Placebo |
69 |
14.5 |
11.5 |
Figure 1 presents the proportion of patients (X axis) whose percentage reduction from baseline in complex partial seizure rates was at least as great as that indicated on the Y axis in the adjunctive therapy study. A positive percent reduction indicates an improvement (i.e., a decrease in seizure frequency), while a negative percent reduction indicates worsening. Thus, in a display of this type, the curve for an effective treatment is shifted to the left of the curve for placebo. This figure shows that the proportion of patients achieving any particular level of improvement was consistently higher for valproate than for placebo. For example, 45% of patients treated with valproate had a ≥ 50% reduction in complex partial seizure rate compared to 23% of patients treated with placebo.
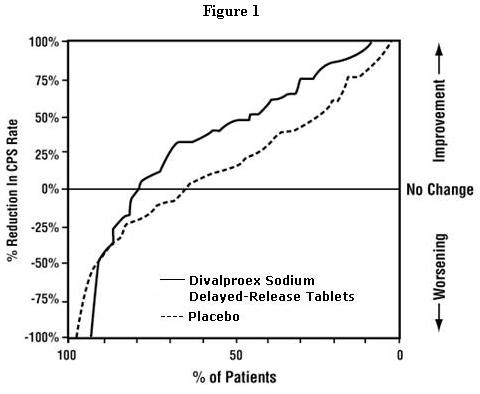
The second study assessed the capacity of valproate to reduce the incidence of CPS when administered as the sole AED. The study compared the incidence of CPS among patients randomized to either a high or low dose treatment arm. Patients qualified for entry into the randomized comparison phase of this study only if
- they continued to experience 2 or more CPS per 4 weeks during an 8 to 12 week long period of monotherapy with adequate doses of an AED (i.e., phenytoin, carbamazepine, phenobarbital, or primidone) and 2) they made a successful transition over a two week interval to valproate. Patients entering the randomized phase were then brought to their assigned target dose, gradually tapered off their concomitant AED and followed for an interval as long as 22 weeks. Less than 50% of the patients randomized, however, completed the study. In patients converted to divalproex sodium delayed-release tablets monotherapy, the mean total valproate concentrations during monotherapy were 71 and 123 mcg/mL in the low dose and high dose groups, respectively.
The following table presents the findings for all patients randomized who had at least one post-randomization assessment.
Table 10. Monotherapy Study Median Incidence of CPS per 8 Weeks
| |||
|
** Treatment** |
Number |
Baseline |
Randomized |
|
High dose Valproate |
131 |
13.2 |
10.7* |
|
Low dose Valproate |
134 |
14.2 |
13.8 |
Figure 2 presents the proportion of patients (X axis) whose percentage reduction from baseline in complex partial seizure rates was at least as great as that indicated on the Y axis in the monotherapy study. A positive percent reduction indicates an improvement (i.e., a decrease in seizure frequency), while a negative percent reduction indicates worsening. Thus, in a display of this type, the curve for a more effective treatment is shifted to the left of the curve for a less effective treatment. This figure shows that the proportion of patients achieving any particular level of reduction was consistently higher for high dose valproate than for low dose valproate. For example, when switching from carbamazepine, phenytoin, phenobarbital or primidone monotherapy to high dose valproate monotherapy, 63% of patients experienced no change or a reduction in complex partial seizure rates compared to 54% of patients receiving low dose valproate.
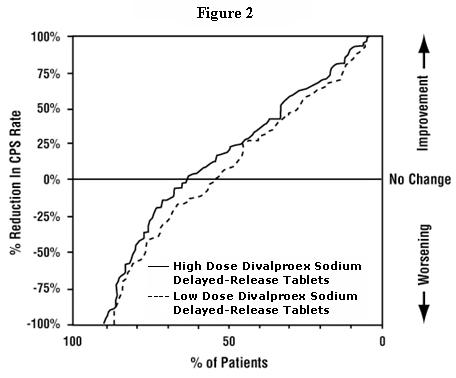
Information on pediatric studies is presented in section 8.
14.3 Migraine
The results of a multicenter, randomized, double-blind, placebo-controlled, parallel-group clinical trial demonstrated the effectiveness of divalproex sodium extended-release tablets in the prophylactic treatment of migraine headache. This trial recruited patients with a history of migraine headaches with or without aura occurring on average twice or more a month for the preceding three months. Patients with cluster or chronic daily headaches were excluded. Women of childbearing potential were allowed in the trial if they were deemed to be practicing an effective method of contraception.
Patients who experienced ≥ 2 migraine headaches in the 4-week baseline period were randomized in a 1:1 ratio to divalproex sodium extended-release tablets or placebo and treated for 12 weeks. Patients initiated treatment on 500 mg once daily for one week, and were then increased to 1,000 mg once daily with an option to permanently decrease the dose back to 500 mg once daily during the second week of treatment if intolerance occurred. Ninety-eight of 114 divalproex sodium extended-release tablets-treated patients (86%) and 100 of 110 placebo-treated patients (91%) treated at least two weeks maintained the 1,000 mg once daily dose for the duration of their treatment periods. Treatment outcome was assessed on the basis of reduction in 4-week migraine headache rate in the treatment period compared to the baseline period.
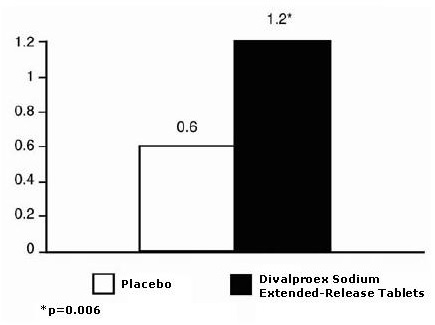
Patients (50 male, 187 female) ranging in age from 16 to 69 were treated with divalproex sodium extended-release tablets (N=122) or placebo (N=115). Four patients were below the age of 18 and 3 were above the age of 65. Two hundred and two patients (101 in each treatment group) completed the treatment period. The mean reduction in 4-week migraine headache rate was 1.2 from a baseline mean of 4.4 in the divalproex sodium extended-release tablets group, versus 0.6 from a baseline mean of 4.2 in the placebo group. The treatment difference was statistically significant (see Figure 3).
Figure 3 Mean Reduction In 4-Week Migraine Headache Rates
SPL MEDGUIDE SECTION
|
MEDICATION GUIDE Divalproex Sodium Extended-Release Tablets USP |
|
What is the most important information I should know about divalproex sodium
extended-release tablets? Call your healthcare provider right away if you get any of the following symptoms:
In some cases, liver damage may continue even though the medicine is stopped. Your healthcare provider will do blood tests to check your liver before and during treatment with divalproex sodium extended-release tablets. 2.Divalproex sodium extended-release tablets may harm your unborn baby.
3.** Swelling (Inflammation) and bleeding (hemorrhaging) of your pancreas that can cause death.** Call your healthcare provider right away if you have any of these symptoms:
4.** Like other antiepileptic drugs, divalproex sodium extended-release
tablets may cause suicidal thoughts or actions in a very small number of
people, about 1 in 500.**
How can I watch for early symptoms of suicidal thoughts and actions?
Call your healthcare provider between visits as needed, especially if you are worried about symptoms. Suicidal thoughts or actions can be caused by things other than medicines. If you have suicidal thoughts or actions, your healthcare provider may check for other causes. |
|
What is divalproex sodium extended-release tablets?
Divalproex sodium extended-release tablets are also used to prevent migraine headaches. Divalproex sodium extended-release tablets are also used to treat acute manic or mixed episodes associated with bipolar disorder with or without psychotic features. |
|
Do not take divalproex sodium extended-release tablets if you:
|
|
Before taking divalproex sodium extended-release tablets, tell your healthcare provider about all of your medical conditions including if you:
**Tell your healthcare provider about all the medicines you take,**including prescription and over-the-counter medicines, vitamins, and herbal supplements. Divalproex sodium extended-release tablets may affect the way other medicines work, and other medicines may affect how divalproex sodium extended-release tablets works. Using divalproex sodium extended-release tablets with other medicines can cause serious side effects.Do notstart or stop other medicines without talking to your healthcare provider. Especially tell your healthcare provider if you take:
You can ask your healthcare provider or pharmacist for a list of these medicines if you are not sure. Know the medicines you take. Keep a list of them and show it to your healthcare provider and pharmacist each time you get a new medicine. |
|
How should I take divalproex sodium extended-release tablets?
|
|
**What should I avoid while taking divalproex sodium extended-release tablets? ** *Do notdrink alcohol while taking divalproex sodium extended-release tablets. Divalproex sodium extended-release tablets and alcohol can affect each other causing side effects such as sleepiness and dizziness.
|
|
What are the possible side effects of divalproex sodium extended-release tablets? **Call your healthcare provider right away if you have any of the symptoms
listed below.**Your healthcare provider may do additional tests before and
during your treatment with divalproex sodium extended-release tablets. Your
healthcare provider may reduce your dose, temporarily stop, or permanently
stop treatment if you have certain side effects.
o bruising or red or purple spots on your skin ***increased ammonia levels in your blood.**High ammonia levels can seriously affect your mental activities, slow your alertness, make you feel tired, or cause vomiting (encephalopathy). This has happened when divalproex sodium extended-release tablets are taken alone or with a medicine called topiramate. Call your health care provider if you have any of these symptoms. ***low body temperature (hypothermia).**A drop in your body temperature to less than 95°F can happen during treatment with divalproex sodium extended-release tablets. Call your healthcare provider if you have any of the following symptoms: o feeling tired ***severe multiorgan reactions.**Treatment with divalproex sodium extended-release tablets may cause severe multiorgan reactions that can be life-threatening or may lead to death. Stop taking divalproex sodium extended-release tablets, and contact your healthcare provider or get medical help right away if you develop any of these symptoms of a severe skin reaction: o fever ***drowsiness or sleepiness in the elderly.**This extreme drowsiness may cause you to eat or drink less than you normally would. Tell your healthcare provider if you are not able to eat or drink as you normally do. Your healthcare provider may start you at a lower dose of divalproex sodium extended-release tablets. ***medicine residue in your stool.**Tell your healthcare provider if you have or think you may have medicine residue in your stool. The common side effects of divalproex sodium extended-release tablets include:
These are not all of the possible side effects ofdivalproex sodium extended-release tablets. ** Call your doctor for medical advice about side effects. You may report side effects to FDA at 1-800-FDA-1088.** |
|
How should I store divalproex sodium extended-release tablets?
Keep divalproex sodium extended-release tablets and all medicines out of the reach of children. |
|
General information about the safe and effective use of divalproex sodium
extended-release tablets You can ask your pharmacist or healthcare provider for information about divalproex sodium extended-release tablets that is written for health professionals. |
|
What are the ingredients in divalproex sodium extended-release tablets? Inactive ingredients: FD&C Blue #1, FD&C Blue #2, hypromellose, mannitol, polyacrylate dispersion 40 percent, polyethylene glycol, pregelatinised starch (maize), propylene glycol, shellac glaze in ethanol, silicified microcrystalline cellulose, silicon dioxide, titanium dioxide, and triacetin. In addition, 500 mg tablets contain iron oxide black, iron oxide yellow, and polydextrose. Brands listed are the trademarks of their respective owners and not trademarks of Aurobindo Pharma Limited. Distributed by: Manufactured by: For more information, call Aurobindo Pharma USA, Inc. at 1-866-850-2876. |
This Medication Guide has been approved by the U.S. Food and Drug Administration.
Revised: 05/2023
Dosage & Administration Section
2 DOSAGE AND ADMINISTRATION
Divalproex sodium extended-release tablets are an extended-release product intended for once-a-day oral administration. Divalproex sodium extended- release tablets should be swallowed whole and should not be crushed or chewed.
2.1 Mania
Divalproex sodium extended-release tablets are administered orally. The recommended initial dose is 25 mg/kg/day given once daily. The dose should be increased as rapidly as possible to achieve the lowest therapeutic dose which produces the desired clinical effect or the desired range of plasma concentrations. In a placebo-controlled clinical trial of acute mania or mixed type, patients were dosed to a clinical response with a trough plasma concentration between 85 and 125 mcg/mL. The maximum recommended dosage is 60 mg/kg/day.
There is no body of evidence available from controlled trials to guide a clinician in the longer term management of a patient who improves during divalproex sodium extended-release tablets treatment of an acute manic episode. While it is generally agreed that pharmacological treatment beyond an acute response in mania is desirable, both for maintenance of the initial response and for prevention of new manic episodes, there are no data to support the benefits of divalproex sodium extended-release tablets in such longer-term treatment (i.e., beyond 3 weeks).
2.2 Epilepsy
Divalproex sodium extended-release tablets are administered orally, and must be swallowed whole. As divalproex sodium extended-release tablets dosage is titrated upward, concentrations of clonazepam, diazepam, ethosuximide, lamotrigine, tolbutamide, phenobarbital, carbamazepine, and/or phenytoin may be affected [see Drug Interactions (7.2)].****
** Complex Partial Seizures**
****For adults and children 10 years of age or older.
Monotherapy (Initial Therapy)
Divalproex sodium extended-release tablets have not been systematically studied as initial therapy. Patients should initiate therapy at 10 to 15 mg/kg/day. The dosage should be increased by 5 to 10 mg/kg/week to achieve optimal clinical response. Ordinarily, optimal clinical response is achieved at daily doses below 60 mg/kg/day. If satisfactory clinical response has not been achieved, plasma levels should be measured to determine whether or not they are in the usually accepted therapeutic range (50 to 100 mcg/mL). No recommendation regarding the safety of valproate for use at doses above 60 mg/kg/day can be made.
The probability of thrombocytopenia increases significantly at total trough valproate plasma concentrations above 110 mcg/mL in females and 135 mcg/mL in males. The benefit of improved seizure control with higher doses should be weighed against the possibility of a greater incidence of adverse reactions.
Conversion to Monotherapy
Patients should initiate therapy at 10 to 15 mg/kg/day. The dosage should be increased by 5 to 10 mg/kg/week to achieve optimal clinical response. Ordinarily, optimal clinical response is achieved at daily doses below 60 mg/kg/day. If satisfactory clinical response has not been achieved, plasma levels should be measured to determine whether or not they are in the usually accepted therapeutic range (50 to 100 mcg/mL). No recommendation regarding the safety of valproate for use at doses above 60 mg/kg/day can be made.
Concomitant antiepilepsy drug (AED) dosage can ordinarily be reduced by approximately 25% every 2 weeks. This reduction may be started at initiation of divalproex sodium extended-release tablets therapy, or delayed by 1 to 2 weeks if there is a concern that seizures are likely to occur with a reduction. The speed and duration of withdrawal of the concomitant AED can be highly variable, and patients should be monitored closely during this period for increased seizure frequency.
Adjunctive Therapy
Divalproex sodium extended-release tablets may be added to the patient's regimen at a dosage of 10 to 15 mg/kg/day. The dosage may be increased by 5 to 10 mg/kg/week to achieve optimal clinical response. Ordinarily, optimal clinical response is achieved at daily doses below 60 mg/kg/day. If satisfactory clinical response has not been achieved, plasma levels should be measured to determine whether or not they are in the usually accepted therapeutic range (50 to 100 mcg/mL). No recommendation regarding the safety of valproate for use at doses above 60 mg/kg/day can be made.
In a study of adjunctive therapy for complex partial seizures in which patients were receiving either carbamazepine or phenytoin in addition to valproate, no adjustment of carbamazepine or phenytoin dosage was needed [see Clinical Studies (14.2)]. However, since valproate may interact with these or other concurrently administered AEDs as well as other drugs, periodic plasma concentration determinations of concomitant AEDs are recommended during the early course of therapy [see Drug Interactions (7)].
** Simple and Complex Absence Seizures**
****The recommended initial dose is 15 mg/kg/day, increasing at one week intervals by 5 to 10 mg/kg/day until seizures are controlled or side effects preclude further increases. The maximum recommended dosage is 60 mg/kg/day.
A good correlation has not been established between daily dose, serum concentrations, and therapeutic effect. However, therapeutic valproate serum concentration for most patients with absence seizures is considered to range from 50 to 100 mcg/mL. Some patients may be controlled with lower or higher serum concentrations [see Clinical Pharmacology (12.3)].
As divalproex sodium extended-release tablets dosage is titrated upward, blood concentrations of phenobarbital and/or phenytoin may be affected [see Drug Interactions (7.2)].
Antiepilepsy drugs should not be abruptly discontinued in patients in whom the drug is administered to prevent major seizures because of the strong possibility of precipitating status epilepticus with attendant hypoxia and threat to life.
2.3 Migraine
Divalproex sodium extended-release tablets are indicated for prophylaxis of migraine headaches in adults.
The recommended starting dose is 500 mg once daily for 1 week, thereafter increasing to 1,000 mg once daily. Although doses other than 1,000 mg once daily of divalproex sodium extended-release tablets have not been evaluated in patients with migraine, the effective dose range of divalproex sodium delayed- release tablets in these patients is 500 to 1,000 mg/day. As with other valproate products, doses of divalproex sodium extended-release tablets should be individualized and dose adjustment may be necessary. If a patient requires smaller dose adjustments than that available with divalproex sodium extended- release tablets, divalproex sodium delayed-release tablets should be used instead.
2.4 Conversion from Divalproex Sodium Delayed-Release Tablets to Divalproex
Sodium Extended-Release Tablets
In adult patients and pediatric patients 10 years of age or older with epilepsy previously receiving divalproex sodium delayed-release tablets, divalproex sodium extended-release tablets should be administered once-daily using a dose 8 to 20% higher than the total daily dose of divalproex sodium delayed-release tablets (Table 1). For patients whose divalproex sodium delayed-release tablets total daily dose cannot be directly converted to divalproex sodium extended-release tablets, consideration may be given at the clinician’s discretion to increase the patient’s divalproex sodium delayed- release tablets total daily dose to the next higher dosage before converting to the appropriate total daily dose of divalproex sodium extended-release tablets.
Table 1. Dose Conversion
| |
|
Divalproex Sodium Delayed-Release Tablets |
Divalproex Sodium Extended-Release Tablets |
|
Total Daily Dose (mg) |
(mg) |
|
500* to 625 |
750 |
|
750* to 875 |
1,000 |
|
1,000* to 1,125 |
1,250 |
|
1,250 to 1,375 |
1,500 |
|
1,500 to 1,625 |
1,750 |
|
1,750 |
2,000 |
|
1,875 to 2,000 |
2,250 |
|
2,125 to 2,250 |
2,500 |
|
2,375 |
2,750 |
|
2,500 to 2,750 |
3,000 |
|
2,875 |
3,250 |
|
3,000 to 3,125 |
3,500 |
There is insufficient data to allow a conversion factor recommendation for patients with divalproex sodium delayed-release tablets doses above 3,125 mg/day. Plasma valproate Cmin concentrations for divalproex sodium extended- release tablets on average are equivalent to divalproex sodium delayed-release tablets, but may vary across patients after conversion. If satisfactory clinical response has not been achieved, plasma levels should be measured to determine whether or not they are in the usually accepted therapeutic range (50 to 100 mcg/mL) [see Clinical Pharmacology (12.2)].
2.5 General Dosing Advice
Dosing in Elderly Patients
Due to a decrease in unbound clearance of valproate and possibly a greater sensitivity to somnolence in the elderly, the starting dose should be reduced in these patients. Starting doses in the elderly lower than 250 mg can only be achieved by the use of divalproex sodium delayed-release tablets. Dosage should be increased more slowly and with regular monitoring for fluid and nutritional intake, dehydration, somnolence, and other adverse reactions. Dose reductions or discontinuation of valproate should be considered in patients with decreased food or fluid intake and in patients with excessive somnolence. The ultimate therapeutic dose should be achieved on the basis of both tolerability and clinical response [see Warnings and Precautions (5.14), Use in Specific Populations (8.5), and Clinical Pharmacology (12.3)].
Dose-Related Adverse Reactions
The frequency of adverse effects (particularly elevated liver enzymes and thrombocytopenia) may be dose-related. The probability of thrombocytopenia appears to increase significantly at total valproate concentrations of ≥ 110 mcg/mL (females) or ≥ 135 mcg/mL (males) [see Warnings and Precautions (5.8)]. The benefit of improved therapeutic effect with higher doses should be weighed against the possibility of a greater incidence of adverse reactions.
G.I. Irritation
Patients who experience G.I. irritation may benefit from administration of the drug with food or by slowly building up the dose from an initial low level.
Compliance
Patients should be informed to take divalproex sodium extended-release tablets every day as prescribed. If a dose is missed it should be taken as soon as possible, unless it is almost time for the next dose. If a dose is skipped, the patient should not double the next dose.
2.6 Dosing in Patients Taking Rufinamide
Patients stabilized on rufinamide before being prescribed valproate should begin valproate therapy at a low dose, and titrate to a clinically effective dose [see Drug Interactions (7.2)].
- Divalproex sodium extended-release tablets are intended for once-a-day oral administration. Divalproex sodium extended-release tablets should be swallowed whole and should not be crushed or chewed (2.1, 2.2).
- Mania: Initial dose is 25 mg/kg/day, increasing as rapidly as possible to achieve therapeutic response or desired plasma level (2.1). The maximum recommended dosage is 60 mg/kg/day (2.1, 2.2).
- Complex Partial Seizures: Start at 10 to 15 mg/kg/day, increasing at 1 week intervals by 5 to 10 mg/kg/day to achieve optimal clinical response; if response is not satisfactory, check valproate plasma level; see full prescribing information for conversion to monotherapy (2.2). The maximum recommended dosage is 60 mg/kg/day (2.1, 2.2).
- Absence Seizures: Start at 15 mg/kg/day, increasing at 1 week intervals by 5 to 10 mg/kg/day until seizure control or limiting side effects (2.2). The maximum recommended dosage is 60 mg/kg/day (2.1, 2.2).
- Migraine: The recommended starting dose is 500 mg/day for 1 week, thereafter increasing to 1,000 mg/day (2.3).