
Skytrofa
These highlights do not include all the information needed to use SKYTROFA safely and effectively. See full prescribing information for SKYTROFA. SKYTROFA (lonapegsomatropin-tcgd) for injection, for subcutaneous use Initial U.S. Approval: 2021
9229f871-bc61-4ddc-8122-d126d40e36ac
HUMAN PRESCRIPTION DRUG LABEL
Sep 4, 2025
Ascendis Pharma Endocrinology, Inc.
DUNS: 118185491
Products 10
Detailed information about drug products covered under this FDA approval, including NDC codes, dosage forms, ingredients, and administration routes.
Lonapegsomatropin-tcgd
Product Details
FDA regulatory identification and product classification information
FDA Identifiers
Product Classification
Product Specifications
INGREDIENTS (1)
Lonapegsomatropin-tcgd
Product Details
FDA regulatory identification and product classification information
FDA Identifiers
Product Classification
Product Specifications
INGREDIENTS (1)
Lonapegsomatropin-tcgd
Product Details
FDA regulatory identification and product classification information
FDA Identifiers
Product Classification
Product Specifications
INGREDIENTS (1)
Lonapegsomatropin-tcgd
Product Details
FDA regulatory identification and product classification information
FDA Identifiers
Product Classification
Product Specifications
INGREDIENTS (1)
Lonapegsomatropin-tcgd
Product Details
FDA regulatory identification and product classification information
FDA Identifiers
Product Classification
Product Specifications
INGREDIENTS (1)
Lonapegsomatropin-tcgd
Product Details
FDA regulatory identification and product classification information
FDA Identifiers
Product Classification
Product Specifications
INGREDIENTS (1)
Lonapegsomatropin-tcgd
Product Details
FDA regulatory identification and product classification information
FDA Identifiers
Product Classification
Product Specifications
INGREDIENTS (1)
Lonapegsomatropin-tcgd
Product Details
FDA regulatory identification and product classification information
FDA Identifiers
Product Classification
Product Specifications
INGREDIENTS (1)
Lonapegsomatropin-tcgd
Product Details
FDA regulatory identification and product classification information
FDA Identifiers
Product Classification
Product Specifications
INGREDIENTS (1)
Lonapegsomatropin-tcgd
Product Details
FDA regulatory identification and product classification information
FDA Identifiers
Product Classification
Product Specifications
INGREDIENTS (1)
Drug Labeling Information
PACKAGE LABEL.PRINCIPAL DISPLAY PANEL
PRINCIPAL DISPLAY PANEL - 2.5 mg Cartridge Blister Pack Box
Skytrofa®
(lonapegsomatropin-tcgd)
for injection
2.5 mg
Box with:
4 single-dose cartridges and
6 sterile injection needles
OPEN HERE
127876
NDC 73362-016-01
GTIN: 00373362016010
LOT NNNNNNNN
Expiration YYYY-MM
SN NNNNNNNNNNNN

INDICATIONS & USAGE SECTION
1 INDICATIONS AND USAGE
SKYTROFA (lonapegsomatropin-tcgd) is a human growth hormone indicated for the:
- Treatment of pediatric patients 1 year and older who weigh at least 11.5 kg and have growth failure due to inadequate secretion of endogenous growth hormone (GH).
- Replacement of endogenous growth hormone in adults with growth hormone deficiency (GHD).
SKYTROFA is a human growth hormone indicated for:
- Pediatric Patients: Treatment of pediatric patients 1 year and older who weigh at least 11.5 kg and have growth failure due to inadequate secretion of endogenous growth hormone (GH) (1).
- Adults: Replacement of endogenous growth hormone in adults with growth hormone deficiency (GHD) (1).
CONTRAINDICATIONS SECTION
4 CONTRAINDICATIONS
SKYTROFA is contraindicated in patients with:
- Acute critical illness after open heart surgery, abdominal surgery or multiple accidental trauma, or those with acute respiratory failure due to the risk of increased mortality with use of pharmacologic doses of somatropin [see Warnings and Precautions (5.1)].
- Hypersensitivity to somatropin or any of the excipients in SKYTROFA. Severe systemic hypersensitivity reactions, including anaphylactic reactions and angioedema, have been reported [see Warnings and Precautions (5.2)].
- Pediatric patients with closed epiphyses.
- Active malignancy due to the risk of malignancy progression [see Warnings and Precautions (5.3)].
- Active proliferative or severe non-proliferative diabetic retinopathy because treatment with somatropin may worsen this condition.
- Pediatric patients with Prader-Willi syndrome who are severely obese, have a history of upper airway obstruction or sleep apnea or have severe respiratory impairment due to the risk of sudden death [see Warnings and Precautions (5.13)].
- Acute critical illness (4)
- Hypersensitivity to somatropin or any of the excipients in SKYTROFA (4)
- Children with closed epiphyses (4)
- Active malignancy (4)
- Active proliferative or severe non-proliferative diabetic retinopathy (4)
- Children with Prader-Willi syndrome who are severely obese or have severe respiratory impairment due to risk of sudden death (4)
WARNINGS AND PRECAUTIONS SECTION
5 WARNINGS AND PRECAUTIONS
5.1 Increased Mortality in Patients With Acute Critical Illness
Increased mortality in patients with acute critical illness due to complications following open heart surgery, abdominal surgery or multiple accidental trauma, or those with acute respiratory failure has been reported after treatment with pharmacologic doses of somatropin [see Contraindications (4)].
The safety of continuing SKYTROFA treatment in patients receiving replacement doses for the approved indication who concurrently develop these illnesses has not been established.
5.2 Severe Hypersensitivity
Severe systemic hypersensitivity reactions, including anaphylactic reactions and angioedema have been reported with postmarketing use of somatropin products, including SKYTROFA. Inform patients and/or caregivers that such reactions are possible, and that prompt medical attention should be sought if an allergic reaction occurs. SKYTROFA is contraindicated in patients with known hypersensitivity to somatropin or any of the excipients in SKYTROFA.
5.3 Increased Risk of Neoplasms
Active Malignancy
There is an increased risk of malignancy progression with somatropin treatment in patients with active malignancy [see Contraindications (4)]. Any preexisting malignancy should be inactive, and its treatment should be completed prior to instituting therapy with SKYTROFA. Discontinue SKYTROFA if there is evidence of recurrent malignancy.
Risk of Second Neoplasm in Pediatric Patients
In childhood cancer survivors who were treated with radiation to the brain/head for their first neoplasm and who developed subsequent growth hormone deficiency (GHD) and were treated with somatropin, an increased risk of a second neoplasm has been reported. Intracranial tumors, in particular meningiomas, were the most common of these second neoplasms. Monitor all patients with a history of GHD secondary to an intracranial neoplasm while on somatropin therapy for progression or recurrence of the tumor.
New Malignancy During Treatment
Because children with certain rare genetic causes of short stature have an increased risk of developing malignancies, thoroughly consider the risks and benefits of starting somatropin in these patients. If treatment with somatropin is initiated, carefully monitor these patients for development of neoplasms.
Monitor patients on SKYTROFA therapy carefully for increased growth or potential malignant changes of preexisting nevi. Advise patients/caregivers to report marked changes in behavior, onset of headaches, vision disturbances and/or changes in skin pigmentation or changes in the appearance of preexisting nevi.
5.4 Glucose Intolerance and Diabetes Mellitus
Treatment with somatropin may decrease insulin sensitivity, particularly at higher doses. Previously undiagnosed impaired glucose tolerance and overt type 2 diabetes mellitus may be unmasked. Monitor glucose levels in all patients receiving SKYTROFA, especially in those with risk factors for type 2 diabetes mellitus, such as obesity or a family history of type 2 diabetes mellitus. When initiating SKYTROFA, monitor closely patients with preexisting type 1 or type 2 diabetes mellitus or impaired glucose tolerance and adjust the doses of antihyperglycemic drugs as needed.
5.5 Intracranial Hypertension
Intracranial hypertension (IH) with papilledema, visual changes, headache, nausea, and/or vomiting has been reported in a small number of patients treated with somatropin. Symptoms usually occurred within 8 weeks after the initiation of somatropin. In all reported cases, IH-associated signs and symptoms resolved rapidly after cessation of therapy or a reduction of the somatropin dose. To exclude preexisting papilledema, perform fundoscopic examination before initiating treatment with SKYTROFA, and reassess periodically thereafter. If papilledema is observed by fundoscopy, stop somatropin treatment. If somatropin-induced IH is confirmed, restart treatment with SKYTROFA at a lower dose after IH-associated signs and symptoms have resolved.
5.6 Fluid Retention
Fluid retention during somatropin therapy may occur. Clinical manifestations of fluid retention (e.g., edema, arthralgia, myalgia, nerve compression syndromes, including carpal tunnel syndrome/paresthesia) are usually transient and dose dependent.
5.7 Hypoadrenalism
Patients receiving somatropin therapy who have or are at risk for pituitary hormone deficiency(s) may be at risk for reduced serum cortisol levels and/or unmasking of central (secondary) hypoadrenalism. In addition, patients treated with glucocorticoid replacement for previously diagnosed hypoadrenalism may require an increase in their maintenance or stress doses following initiation of SKYTROFA therapy. Monitor patients for reduced serum cortisol levels and/or need for glucocorticoid dose increases in patients with known hypoadrenalism [see Drug Interactions (7)].
5.8 Hypothyroidism
Undiagnosed or untreated hypothyroidism may prevent optimal response to SKYTROFA. In patients with GHD, central (secondary) hypothyroidism may first become evident or worsen during SKYTROFA treatment. Therefore, perform periodic thyroid function tests in patients and initiate or appropriately adjust thyroid hormone replacement therapy when indicated.
5.9 Slipped Capital Femoral Epiphysis in Pediatric Patients
Slipped capital femoral epiphysis may occur more frequently in patients undergoing rapid growth. Slipped capital femoral epiphysis may lead to osteonecrosis. Cases of slipped capital femoral epiphysis with or without osteonecrosis have been reported in pediatric patients with short stature receiving somatropin. Evaluate pediatric patients receiving SKYTROFA with the onset of a limp or complaints of persistent hip or knee pain for slipped capital femoral epiphysis and osteonecrosis and manage accordingly.
5.10 Progression of Preexisting Scoliosis in Pediatric Patients
Somatropin increases growth rate in pediatric patients, and progression of existing scoliosis can occur in patients who experience rapid growth. Somatropin has not been shown to increase the occurrence of scoliosis. Monitor patients with a history of scoliosis for disease progression.
5.11 Pancreatitis
Pancreatitis has been reported in pediatric patients receiving somatropin. The risk may be greater in pediatric patients than adults. Consider pancreatitis in patients who develop persistent severe abdominal pain.
5.12 Lipoatrophy
When SKYTROFA is administered subcutaneously at the same site over a long period of time, lipoatrophy may result. Rotate injection sites when administering SKYTROFA to reduce this risk [see Dosage and Administration (2.7)].
5.13 Sudden Death in Pediatric Patients With Prader-Willi Syndrome
There have been reports of fatalities after initiating therapy with somatropin in pediatric patients with Prader-Willi syndrome who had one or more of the following risk factors: severe obesity, history of upper airway obstruction or sleep apnea, or unidentified respiratory infection. Male patients with one or more of these factors may be at greater risk than females. SKYTROFA is not indicated for the treatment of pediatric patients who have growth failure due to genetically confirmed Prader-Willi syndrome.
5.14 Laboratory Tests
Serum levels of alkaline phosphatase and phosphate may increase after SKYTROFA therapy. Serum levels of parathyroid hormone may increase after somatropin treatment. If a patient is found to have abnormal laboratory tests, monitor as appropriate.
- Severe Hypersensitivity: Hypersensitivity reactions, including anaphylaxis and angioedema have occurred. In the event of an allergic reaction, seek prompt medical attention (5.2).
- Increased Risk of Neoplasms: Monitor patients with preexisting tumors for progressions or recurrence. Increased risk of a second neoplasm in childhood cancer survivors treated with somatropin – in particular meningiomas in patients treated with radiation to the head for their first neoplasm (5.3).
- Glucose Intolerance and Diabetes Mellitus: May be unmasked. Periodically monitor glucose levels in all patients. Doses of concurrent antihyperglycemic drugs in diabetics may require adjustment (5.4).
- Intracranial Hypertension: Exclude preexisting papilledema. May develop and is usually reversible after discontinuation or dose reduction (5.5).
- Fluid Retention (i.e., edema, arthralgia, carpal tunnel syndrome): May occur. Reduce dose as necessary (5.6).
- Hypoadrenalism: Monitor patients for reduced serum cortisol levels and/or need for glucocorticoid dose increases in those with known hypoadrenalism (5.7).
- Hypothyroidism: May first become evident or worsen (5.8).
- Slipped Capital Femoral Epiphysis in Pediatric Patients: May develop. Evaluate children with the onset of a limp or persistent hip/knee pain (5.9).
- Progression of Preexisting Scoliosis: May develop (5.10).
- Pancreatitis: Consider pancreatitis in patients with persistent severe abdominal pain (5.11).
ADVERSE REACTIONS SECTION
6 ADVERSE REACTIONS
The following important adverse reactions are described elsewhere in the labeling:
- Increased mortality in patients with acute critical illness [see Warnings and Precautions (5.1)]
- Severe hypersensitivity [see Warnings and Precautions (5.2)]
- Increased risk of neoplasms [see Warnings and Precautions (5.3)]
- Glucose intolerance and diabetes mellitus [see Warnings and Precautions (5.4)]
- Intracranial hypertension [see Warnings and Precautions (5.5)]
- Fluid retention [see Warnings and Precautions (5.6)]
- Hypoadrenalism [see Warnings and Precautions (5.7)]
- Hypothyroidism [see Warnings and Precautions (5.8)]
- Slipped capital femoral epiphysis in pediatric patients [see Warnings and Precautions (5.9)]
- Progression of preexisting scoliosis in pediatric patients [see Warnings and Precautions (5.10)]
- Pancreatitis [see Warnings and Precautions (5.11)]
- Lipoatrophy [see Warnings and Precautions (5.12)]
- Sudden death in pediatric patients with Prader-Willi syndrome [see Warnings and Precautions (5.13)]
6.1 Clinical Trials Experience
Because clinical trials are conducted under widely varying conditions, adverse reaction rates observed in the clinical trials of a drug cannot be directly compared to rates in the clinical studies of another drug and may not reflect the rates observed in clinical practice.
Pediatric Patients with Growth Hormone Deficiency
SKYTROFA was studied in a 52-week, open-label, active-controlled trial in 161 treatment-naïve, prepubertal pediatric patients with growth hormone deficiency (GHD) [see Clinical Studies (14.1)]. The subjects ranged in age from 3.2 to 13.1 years with a mean of 8.5 years. One hundred thirty-two (82%) of the subjects were male and 29 (18%) were female. One subject was Asian, 3 were Black or African American, 152 were Caucasian, and 5 were categorized as "other."
Table 2 shows common adverse reactions that occurred in ≥ 5% of patients treated with SKYTROFA in this trial.
Table 2: Adverse Reactions Occurring in ≥ 5% SKYTROFA-Treated Pediatric Patients and More Frequently Than in Daily Somatropin-Treated Pediatric Patients (52 Weeks of Treatment)|
Adverse reactions |
Daily Somatropin |
SKYTROFA |
|---|---|---|
|
Adverse reactions that are medically related were grouped to a single preferred term. | ||
| ||
|
Infection, viral |
6 (11%) |
16 (15%) |
|
Pyrexia |
5 (9%) |
16 (15%) |
|
Cough |
4 (7%) |
11 (11%) |
|
Nausea and vomiting |
4 (7%) |
11 (11%) |
|
Hemorrhage* |
1 (2%) |
7 (7%) |
|
Diarrhea |
3 (5%) |
6 (6%) |
|
Abdominal pain |
2 (4%) |
6 (6%) |
|
Arthralgia and arthritis† |
1 (2%) |
6 (6%) |
Laboratory Tests
More SKYTROFA-treated patients shifted from normal baseline levels to elevated phosphate and alkaline phosphatase levels at the end of the trial compared to the daily somatropin group (44.2% vs. 30.2% and 19.2% vs. 9.4%, respectively); these laboratory changes occurred intermittently [see Warnings and Precautions (5.14)].
Adults with Growth Hormone Deficiency
SKYTROFA was studied in a 38-week parallel-arm, placebo-controlled (double- blind) and active-controlled (open label) trial in 259 adults with growth hormone deficiency (GHD) [see Clinical Studies (14.2)].
The mean (range) age at enrollment was 43 (23 to 81) years old, with 119 (46%) females (55 on oral estrogen) and 140 (54%) males. One subject was American Indian or Alaska Native, one was Black or African American, 28 were Asian, and 218 were Caucasian.
Table 3 shows adverse reactions that occurred in ≥ 5% of adults treated with SKYTROFA and more frequently than in placebo-treated adults in this trial.
Table 3: Adverse Reactions Occurring in ≥ 5% of SKYTROFA-Treated Adults and More Frequently Than in Placebo-treated Adults (38 Weeks of Treatment)|
Adverse reactions |
Placebo |
SKYTROFA |
|---|---|---|
|
Adverse reactions that are medically related were grouped to a single preferred term. | ||
| ||
|
Edema* |
1 (1%) |
7 (8%) |
|
Central (secondary) hypothyroidism† |
1 (1%) |
6 (7%) |
Laboratory Tests
More SKYTROFA-treated patients shifted from normal or low baseline levels to elevated alkaline phosphatase levels at the end of the trial compared to the placebo group (14% vs. 6%); these laboratory changes were noted with increased frequency as the trial progressed [see Warnings and Precautions (5.14)].
6.2 Postmarketing Experience
The following adverse reactions have been identified during post approval use of somatropin products or SKYTROFA. Because these reactions are reported voluntarily from a population of uncertain size, it is not always possible to reliably estimate their frequency or establish a causal relationship to drug exposure.
- Severe systemic hypersensitivity reactions, including anaphylactic reactions and angioedema
- Musculoskeletal and connective tissue disorders – osteonecrosis in pediatric patients
Pediatric Patients: Most common adverse reactions (≥ 5%): viral infection, pyrexia, cough, nausea and vomiting, hemorrhage, diarrhea, abdominal pain, and arthralgia and arthritis (6.1).
Adults: Most common adverse reaction (≥ 5%): edema peripheral (6.1).
To report SUSPECTED ADVERSE REACTIONS, contact Ascendis Pharma Endocrinology, Inc., at 1-844-442-7236 or FDA at 1-800-FDA-1088 or www.fda.gov/medwatch.
DRUG INTERACTIONS SECTION
7 DRUG INTERACTIONS
Table 4 includes a list of drugs with clinically important drug interactions when administered concomitantly with SKYTROFA and instructions for preventing or managing them.
Table 4: Clinically Important Drug Interactions with SKYTROFA|
Replacement Glucocorticoid Treatment | |
|
Clinical Impact: |
Microsomal enzyme 11β-hydroxysteroid dehydrogenase type 1 (11βHSD-1) is required for conversion of cortisone to its active metabolite, cortisol, in hepatic and adipose tissue. Somatropin inhibits 11βHSD-1. Consequently, individuals with untreated growth hormone deficiency (GHD) have relative increases in 11βHSD-1 and serum cortisol. Initiation of SKYTROFA may result in inhibition of 11βHSD-1 and reduced serum cortisol concentrations. |
|
Intervention: |
Patients treated with glucocorticoid replacement for hypoadrenalism may require an increase in their maintenance or stress doses following initiation of SKYTROFA [see Warnings and Precautions (5.7)] |
|
Examples |
Cortisone acetate and prednisone may be affected more than others because conversion of these drugs to their biologically active metabolites is dependent on the activity of 11βHSD-1. |
|
Pharmacologic Glucocorticoid Therapy and Supraphysiologic Glucocorticoid Treatment | |
|
Clinical Impact: |
Pharmacologic glucocorticoid therapy and supraphysiologic glucocorticoid treatment may attenuate the growth-promoting effects of SKYTROFA in pediatric patients. |
|
Intervention: |
Carefully adjust glucocorticoid replacement dosing in pediatric patients receiving glucocorticoid treatments to avoid both hypoadrenalism and an inhibitory effect on growth. |
|
Cytochrome P450-Metabolized Drugs | |
|
Clinical Impact: |
Limited published data indicate that somatropin treatment increases cytochrome P450 (CYP450)-mediated antipyrine clearance. SKYTROFA may alter the clearance of compounds known to be metabolized by CYP450 liver enzymes. |
|
Intervention: |
Careful monitoring is advisable when SKYTROFA is administered in combination with drugs metabolized by CYP450 liver enzymes. |
|
Oral Estrogen | |
|
Clinical Impact: |
Oral estrogens may reduce the serum insulin-like growth factor-1 (IGF-1) response to SKYTROFA. |
|
Intervention: |
Patients receiving oral estrogen replacement may require higher SKYTROFA dosages. |
|
Insulin and/or Other Antihyperglycemic Agents | |
|
Clinical Impact: |
Treatment with SKYTROFA may decrease insulin sensitivity, particularly at higher doses. |
|
Intervention: |
Patients with diabetes mellitus may require adjustment of their doses of insulin and/or other antihyperglycemic agents [see Warnings and Precautions (5.4)]. |
- Replacement Glucocorticoid Treatment: Patients treated with glucocorticoid for hypoadrenalism may require an increase in their maintenance or stress doses following initiation of SKYTROFA (7).
- Pharmacologic Glucocorticoid Therapy and Supraphysiologic Glucocorticoid Treatment: Adjust glucocorticoid dosing in pediatric patients to avoid both hypoadrenalism and an inhibitory effect on growth (7).
- Cytochrome P450-Metabolized Drugs: SKYTROFA may alter the clearance. Monitor carefully if used with SKYTROFA (7).
- Oral Estrogen: Larger doses of SKYTROFA may be required (7).
- Insulin and/or Other Antihyperglycemic Agents: Dose adjustment of insulin or antihyperglycemic agent may be required (7).
CLINICAL PHARMACOLOGY SECTION
12 CLINICAL PHARMACOLOGY
12.1 Mechanism of Action
SKYTROFA is a pegylated human growth hormone (somatropin) for once-weekly subcutaneous injection [see Clinical Pharmacology (12.3)].
Somatropin binds to the growth hormone (GH) receptor in the cell membrane of target cells resulting in intracellular signal transduction and a host of pharmacodynamic effects. Somatropin has direct tissue and metabolic effects, and indirect effects mediated by insulin-like growth factor-1 (IGF-1), including stimulation of chondrocyte differentiation and proliferation, stimulation of hepatic glucose output, protein synthesis and lipolysis. Somatropin stimulates skeletal growth in pediatric patients with growth hormone deficiency (GHD) as a result of effects on the growth plates (epiphyses) of long bones.
12.2 Pharmacodynamics
Somatropin released from SKYTROFA produces a linear IGF-1 dose response. In pediatric patients with GHD, with a dose change of 0.02 mg/kg/week on average resulting in a mean change in weekly average IGF-1 standard deviation score (SDS) of 0.17. In adults with GHD, a dose change of 1 mg/week results in a mean change in weekly average IGF-1 SDS of 0.7 (range, 0.3 to 1.2).
At steady state, IGF-1 levels peak approximately 2 days post-dose, with the weekly average IGF-1 occurring approximately 4.5 days post-dose. IGF-1 levels are in the normal range for GHD patients for the majority of the week, similar to daily somatropin.
12.3 Pharmacokinetics
Absorption
Following subcutaneous dose administration, SKYTROFA releases fully active somatropin via autocleavage of the TransCon linker that follows first-order kinetics.
In pediatric patients with GHD, following subcutaneous dose administration of 0.24 mg/kg/week SKYTROFA, the observed mean (CV%) steady-state peak serum concentration (Cmax) of lonapegsomatropin-tcgd was 1230 (86.3) ng hGH/mL, and the median time to reach maximum concentrations (Tmax) was 25 hours. For released somatropin, Cmax was 15.2 (83.4) ng/mL with a median Tmax of 12 hours. The mean (CV%) somatropin exposure over the one-week dose interval (area under the curve) was 500 (83.8) h*ng/mL. Following subcutaneous dose administration of 0.24 mg/kg/week SKYTROFA, mean Cmax (CV%) of the methoxypolyethylene glycol carrier was 13.1 (28.1) mcg/mL with a median Tmax of 36 hours.
In adult patients with GHD, following subcutaneous administration of 6.3 mg/week SKYTROFA, the estimated median steady-state Cmax of lonapegsomatropin- tcgd was 69.3 ng hGH/mL, and the Tmax was 31 hours. For released somatropin, the estimated median Cmax was 2.47 ng/mL with a median Tmax of 13 hours. The median somatropin exposure over the one-week dose interval (area under the curve) was 75.4 h*ng/mL. Following subcutaneous dose administration of 6.3 mg/week SKYTROFA, the estimated median Cmax of the methoxypolyethylene glycol carrier was 4.60 mcg/mL with a median Tmax of 34 hours.
No significant accumulation of lonapegsomatropin-tcgd and somatropin following repeat dose administration was observed.
Distribution
In pediatric patients with GHD, the mean (CV%) steady-state apparent volume of distribution of lonapegsomatropin-tcgd after subcutaneous administration of 0.24 mg/kg/week SKYTROFA was 0.13 (109) L/kg.
In adult patients with GHD, the median steady-state apparent volume of distribution of lonapegsomatropin-tcgd after subcutaneous administration of 6.3 mg/week SKYTROFA was 0.13 L/kg.
A similar distribution pattern as observed for daily somatropin is expected once somatropin is released from lonapegsomatropin-tcgd.
Elimination
Metabolism
The metabolism of somatropin involves protein catabolism in both the liver and kidneys. The methoxypolyethylene glycol carrier is cleared by the kidneys.
Excretion
In pediatric patients with GHD, the mean (CV%) lonapegsomatropin-tcgd apparent clearance at steady state was 3.2 (67) mL/h/kg following subcutaneous administration of 0.24 mg/kg/week SKYTROFA with a mean (±SD) observed half- life of 30.7 (±12.7) hours.
The apparent half-life of somatropin released from lonapegsomatropin-tcgd was approximately 25 hours.
Specific Populations
Based on a population pharmacokinetic analysis, age (3.0 to 13.7 years for pediatric patients and 23 to 75 years for adult patients), sex, race, and body weight do not have clinically meaningful effects on pharmacokinetics.
Male and Female Patients — No sex-specific pharmacokinetic studies have been performed with SKYTROFA. The available literature indicates that the pharmacokinetics of somatropin are similar in men and women.
Patients with Renal or Hepatic Impairment — No specific studies have been performed with SKYTROFA.
12.6 Immunogenicity
The observed incidence of anti-drug antibodies is highly dependent on the sensitivity and specificity of the assay. Differences in assay methods preclude meaningful comparisons of the incidence of anti-drug antibodies in the studies described below with the incidence of anti-drug antibodies in other studies, including those of SKYTROFA or other growth hormone products.
Anti-lonapegsomatropin-tcgd antibodies were evaluated in samples collected every 3 months in phase 3 trials in pediatric patients with GHD receiving SKYTROFA. Mean duration of exposure to SKYTROFA was 70.2 weeks. Of the 304 patients with post-baseline assessments, 19 (6.3%) showed detectable binding antibodies to lonapegsomatropin-tcgd. No apparent correlation of anti- lonapegsomatropin-tcgd antibodies to adverse events or loss of efficacy was observed. No neutralizing antibodies to SKYTROFA were detected.
Of the 247 adult patients with GHD, treated with SKYTROFA and with post- baseline assessments, antibodies against lonapegsomatropin-tcgd were detected in 14 patients (5.7%, mean duration of exposure was 47.7 weeks). There was no identified clinically significant effect of anti-lonapegsomatropin-tcgd antibodies on the safety and efficacy of SKYTROFA. No neutralizing antibodies to SKYTROFA were detected.
INFORMATION FOR PATIENTS SECTION
17 PATIENT COUNSELING INFORMATION
Administration Instructions
- Advise patients and/or caregivers to read the accompanying FDA-approved patient labeling SKYTROFA Auto-Injector Instructions for Use (also available at www.Skytrofa.com/IFU) prior to administration. Advise patients and/or caregivers to call the Ascendis Pharma Customer Support toll-free number at 1-844-442-7236 (1-844-44ASCENDIS) for assistance or additional training, if needed.
- Advise patients and/or caregivers to refer to the Instructions for Use that accompanies the SKYTROFA Auto-Injector for complete mixing and administration instructions with illustrations [see Dosage and Administration (2.7)]. Instruct patients and/or caregivers of proper needle disposal and caution against any reuse of needles. An appropriate container for the disposal of used cartridge and needle should be used.
- Advise patients and/or caregivers to administer SKYTROFA once weekly, at any time of day. Advise patients and/or caregivers that doses can be taken 2 days before or 2 days after the scheduled dosing day. Advise patients and/or caregivers to resume once-weekly dosing for the next dose. If more than 2 days have passed from the schedule dosing day, advise patients and/or caregivers to skip the missed dose and take the next dose on the regularly scheduled day. If subsequently changing the regular dosing day to a different day of the week, advise patients and/or caregivers to ensure that at least 5 days will elapse between the last dose and the newly established regular dosing day.
Hypersensitivity Reactions
Advise patients and/or caregivers that severe and/or serious systemic hypersensitivity reactions (anaphylaxis and angioedema) have been reported, and to seek prompt medical attention should an allergic reaction occur [see Warnings and Precautions (5.2)].
Neoplasms
Advise childhood cancer survivors and/or caregivers that individuals treated with brain and/or head radiation are at increased risk of secondary neoplasms and, as a precaution, need to be monitored for recurrence. Advise patients and/or caregivers to report marked changes in behavior, onset of headaches, vision disturbances and/or changes in skin pigmentation or changes in the appearance of preexisting nevi.
Glucose Intolerance/Diabetes Mellitus
Advise patients and/or caregivers that new onset impaired glucose intolerance/type 2 diabetes mellitus or exacerbation of preexisting diabetes mellitus can occur and monitoring of blood glucose during treatment with SKYTROFA may be needed.
Intracranial Hypertension
Advise patients and/or caregivers to report to their healthcare provider any visual changes, headache, and nausea and/or vomiting.
Fluid Retention
Advise patients and/or caregivers that fluid retention during SKYTROFA replacement therapy may occur. Inform patients and/or caregivers of the clinical manifestations of fluid retention (e.g., edema, arthralgia, myalgia, nerve compression syndromes, including carpal tunnel syndrome/paresthesia) and to report to their healthcare provider if any of these signs or symptoms occur during treatment with SKYTROFA.
Hypoadrenalism
Advise patients and/or caregivers that patients who have or who are at risk for pituitary hormone deficiency(s) that hypoadrenalism may develop and to report to their healthcare provider if they experience hyperpigmentation, extreme fatigue, dizziness, weakness, or weight loss.
Hypothyroidism
Advise patients and/or caregivers that undiagnosed/untreated hypothyroidism may prevent an optimal response to SKYTROFA. Advise patients/caregivers that patients may require periodic thyroid function tests.
Pancreatitis
Advise patients and/or caregivers that pancreatitis may develop and to report to their healthcare provider any new onset abdominal pain.
RECENT MAJOR CHANGES SECTION
RECENT MAJOR CHANGES
|
Indications and Usage (1) |
7/2025 |
|
Dosage and Administration (2.2, 2.3, 2.6, 2.7) |
7/2025 |
|
Warnings and Precautions, Slipped Capital Femoral Epiphysis (5.9) |
7/2025 |
|
Warnings and Precautions, Laboratory Tests (5.14) |
7/2025 |
DOSAGE & ADMINISTRATION SECTION
2 DOSAGE AND ADMINISTRATION
2.1 General Dosing Information
- For subcutaneous injection, once weekly.
- Therapy with SKYTROFA should be supervised by a healthcare provider who is experienced in the diagnosis and management of patients with growth hormone deficiency (GHD).
- Perform fundoscopic examination before initiating treatment with SKYTROFA to exclude preexisting papilledema. If papilledema is identified, evaluate the etiology and treat the underlying cause before initiating treatment with SKYTROFA [see Warnings and Precautions (5.5)].
2.2 Recommended Dosage for Pediatric Patients
- The recommended dose of SKYTROFA for treatment-naïve patients and patients switching from another growth hormone product is 0.24 mg/kg body weight, given once weekly.
- Individualize and titrate the dosage of SKYTROFA based on response.
- When changing from daily somatropin therapy to once-weekly SKYTROFA, wait at least 8 hours between the final dose of daily somatropin and the first dose of SKYTROFA.
- When changing from another once-weekly growth hormone therapy to once-weekly SKYTROFA, wait at least 7 days between the final dose of the previous growth hormone therapy and the first dose of SKYTROFA.
- Assess compliance and evaluate other causes of poor growth, such as hypothyroidism, under-nutrition, advanced bone age and antibodies to recombinant human growth hormone if patients experience failure to increase height velocity, particularly during the first year of treatment.
- Patients who were treated with SKYTROFA for GH deficiency in childhood and whose epiphyses are closed should be reevaluated before continuing SKYTROFA.
2.3 Recommended Dosage for Adults
- The recommended starting dose of SKYTROFA in adults with GHD is based on age and concomitant use of oral estrogen [see Dosage Forms and Strengths (3)]. For treatment-naïve patients or for patients switching from another growth hormone product, start SKYTROFA as described below:
- 1.4 mg once weekly for adults 30 to 60 years old, with no oral estrogen intake
- 2.1 mg once weekly for adults under 30 years old, or adults of any age intaking oral estrogen
- 0.7 mg once weekly for adults over 60 years old, with no oral estrogen intake
- When changing from daily somatropin therapy to once-weekly SKYTROFA, wait at least 8 hours between the final dose of daily somatropin and the first dose of SKYTROFA.
- When changing from another once-weekly growth hormone therapy to once-weekly SKYTROFA, wait at least 7 days between the final dose of the previous growth hormone therapy and the first dose of SKYTROFA.
- Increase the dose monthly to a higher strength cartridge based on the clinical response and/or IGF-1 concentration. Draw IGF-1 serum sample 4 to 5 days after the prior dose.
- Decrease the dose to a lower strength cartridge as needed based on adverse reactions or a weekly average IGF-1 concentration above the age- and sex-specific normal range.
- The maximum recommended dose is 6.3 mg once weekly.
2.4 Missed Doses
- Administer a missed dose as soon as possible and not more than 2 days after the missed dose.
- To avoid missed doses, SKYTROFA can be taken 2 days before or 2 days after the scheduled dosing day. Resume once-weekly dosing for the next dose at the previously scheduled dosing day.
- If more than 2 days have passed from the scheduled day, skip the dose and administer the next dose on the regularly scheduled day.
- At least 5 days should elapse between doses.
2.5 Administration Instructions for Pediatric Patients
SKYTROFA is available in 9 cartridges (dosage strengths in somatropin equivalents) for pediatric patients.
Selection of the appropriate cartridge (mg) is based on the prescribed dose (mg/kg) and the patient's body weight (kg) [see Dosage Forms and Strengths (3)].
- If prescribing a dose of 0.24 mg/kg/week and the patient's weight is 11.5 to 100 kg, follow the recommended dosing in Table 1.
- If prescribing a dose other than 0.24 mg/kg/week, calculate the total weekly dose (in mg) and select the appropriate cartridge as follows:
- Total weekly dose (mg) = prescribed weekly dose (mg/kg) × patient's body weight (kg).
- Round the total weekly dose (mg) to the closest cartridge dose while also considering treatment goals and clinical response.
|
Weight (kg) |
Dose (mg) |
|---|---|
|
11.5 – 13.9 |
3 |
|
14 – 16.4 |
3.6 |
|
16.5 – 19.9 |
4.3 |
|
20 – 23.9 |
5.2 |
|
24 – 28.9 |
6.3 |
|
29 – 34.9 |
7.6 |
|
35 – 41.9 |
9.1 |
|
42 – 50.9 |
11 |
|
51 – 60.4 |
13.3 |
|
60.5 – 69.9 |
15.2 (using two cartridges of 7.6 mg each) |
|
70 – 84.9 |
18.2 (using two cartridges of 9.1 mg each) |
|
85 – 100 |
22 (using two cartridges of 11 mg each) |
2.6 Administration Instructions for Adults
SKYTROFA is available in 14 cartridges (dosage strengths in somatropin equivalents) for adults.
Selection of the appropriate cartridge (mg) is based on the prescribed dose (mg/week) [see Dosage Forms and Strengths (3)].
2.7 Preparation and Administration
- The SKYTROFA cartridge has been designed for use only with the SKYTROFA Auto-Injector.
- If refrigerated, the SKYTROFA cartridge must be kept at room temperature for 15 minutes before use.
- The SKYTROFA Auto-Injector provides a fully automated reconstitution of the lyophilized drug product which is followed by a manual mixing step controlled by the device. When the injection needle is inserted into the skin, the device automatically delivers the drug product. The built-in electronics and software assist the user during the entire preparation and injection sequence and provide confirmation that the full dose has been delivered.
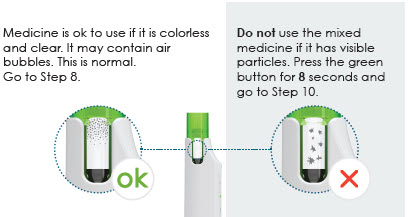
- The mixed solution should be clear and colorless to opalescent. The solution may contain air bubbles and this is acceptable. DO NOT inject if the solution is cloudy or contains particulate matter.
- Use SKYTROFA cartridges within 4 hours after reconstitution. Discard reconstituted SKYTROFA cartridges after 4 hours when stored at room temperature up to 86°F (30°C).
- Inject SKYTROFA subcutaneously into the abdomen, buttock, or thigh. Rotate injection sites between and within regions to reduce the risk of lipoatrophy [see Warnings and Precautions (5.12)].
- Refer to the Instructions for Use for complete administration instructions with illustrations. The instructions can also be found on www.Skytrofa.com/IFU.
- Patients and/or caregivers who will administer SKYTROFA should receive appropriate training and instruction on the proper use of SKYTROFA from their healthcare provider.
- SKYTROFA should be administered subcutaneously once weekly into the abdomen, buttock, or thigh with regular rotation of the injection sites (2.7).
- Pediatric Patients: Recommended dose is 0.24 mg/kg body weight once weekly (2.2).
- Adults: Recommended starting dose is based on age and concomitant use of oral estrogen. Titrate monthly until the desired clinical response and/or weekly average IGF-1 concentration are achieved (2.3).
- See Full Prescribing Information for instructions on preparation and administration of drug (2.5, 2.6, 2.7).
DOSAGE FORMS & STRENGTHS SECTION
3 DOSAGE FORMS AND STRENGTHS
SKYTROFA is a white to off-white lyophilized powder available in a single- dose, dual-chamber, prefilled cartridge containing lonapegsomatropin-tcgd in one chamber and diluent, Water for Injection, in the other chamber and is available in the following strengths:
For injection: 0.7 mg, 1.4 mg, 1.8 mg, 2.1 mg, 2.5 mg, 3 mg, 3.6 mg, 4.3 mg, 5.2 mg, 6.3 mg, 7.6 mg, 9.1 mg, 11 mg, and 13.3 mg.
SKYTROFA is a lyophilized powder available in single-dose, dual-chamber, prefilled cartridges containing lonapegsomatropin-tcgd and diluent, Water for Injection, as follows:
For injection: 0.7 mg, 1.4 mg, 1.8 mg, 2.1 mg, 2.5 mg, 3 mg, 3.6 mg, 4.3 mg, 5.2 mg, 6.3 mg, 7.6 mg, 9.1 mg, 11 mg, and 13.3 mg (3)
OVERDOSAGE SECTION
10 OVERDOSAGE
Acute overdosage may lead initially to hypoglycemia and subsequently to hyperglycemia. Overdose with somatropin may cause fluid retention. Long-term overdosage may result in signs and symptoms of gigantism consistent with the known effects of excess growth hormone.
DESCRIPTION SECTION
11 DESCRIPTION
Lonapegsomatropin-tcgd is a long-acting prodrug of a human growth hormone (somatropin) produced by recombinant DNA technology using E. coli. Lonapegsomatropin-tcgd consists of a parent drug, somatropin, that is conjugated to a methoxypolyethylene glycol carrier (4 × 10 kDa mPEG) via a proprietary TransCon Linker and has a molecular weight of 63 kDa (released somatropin is 22 kDa). In vitro assay confirms the minimum potency of released somatropin is NLT 2.5 IU/mg.
SKYTROFA (lonapegsomatropin-tcgd) for injection is a sterile, preservative- free, white to off-white lyophilized powder available in a single-dose, dual- chamber, prefilled cartridge containing lonapegsomatropin-tcgd in one chamber and the diluent, Water for Injection, in the other chamber. SKYTROFA prefilled cartridge must be used with SKYTROFA Auto-Injector to provide an automatic mixing step for reconstitution prior to subcutaneous use.
After reconstitution, each prefilled cartridge delivers:
- 0.327 mL containing 0.7 mg lonapegsomatropin-tcgd, succinic acid (0.39 mg), trehalose dihydrate (29.8 mg) and tromethamine for pH adjustment to 5.0.
- 0.327 mL containing 1.4 mg lonapegsomatropin-tcgd, succinic acid (0.39 mg) trehalose dihydrate (29.1 mg) and tromethamine for pH adjustment to 5.0.
- 0.327 mL containing 1.8 mg lonapegsomatropin-tcgd, succinic acid (0.39 mg) trehalose dihydrate (28.8 mg) and tromethamine for pH adjustment to 5.0.
- 0.327 mL containing 2.1 mg lonapegsomatropin-tcgd, succinic acid (0.39 mg) trehalose dihydrate (28.4 mg) and tromethamine for pH adjustment to 5.0.
- 0.327 mL containing 2.5 mg lonapegsomatropin-tcgd, succinic acid (0.39 mg) trehalose dihydrate (28.1 mg) and tromethamine for pH adjustment to 5.0.
- 0.273 mL containing 3 mg lonapegsomatropin-tcgd, succinic acid (0.32 mg), trehalose dihydrate (22.7 mg) and tromethamine for pH adjustment to 5.0.
- 0.327 mL containing 3.6 mg lonapegsomatropin-tcgd, succinic acid (0.39 mg), trehalose dihydrate (27.1 mg) and tromethamine for pH adjustment to 5.0.
- 0.391 mL containing 4.3 mg lonapegsomatropin-tcgd, succinic acid (0.46 mg) and trehalose dihydrate (32.5 mg) and tromethamine for pH adjustment to 5.0.
- 0.473 mL containing 5.2 mg lonapegsomatropin-tcgd, succinic acid (0.56 mg) and trehalose dihydrate (39.3 mg) and tromethamine for pH adjustment to 5.0.
- 0.286 mL containing 6.3 mg lonapegsomatropin-tcgd, succinic acid (0.34 mg) and trehalose dihydrate (21.2 mg) and tromethamine for pH adjustment to 5.0.
- 0.345 mL containing 7.6 mg lonapegsomatropin-tcgd, succinic acid (0.41 mg) and trehalose dihydrate (25.5 mg) and tromethamine for pH adjustment to 5.0.
- 0.414 mL containing 9.1 mg lonapegsomatropin-tcgd, succinic acid (0.49 mg) and trehalose dihydrate (30.6 mg) and tromethamine for pH adjustment to 5.0.
- 0.5 mL containing 11 mg lonapegsomatropin-tcgd, succinic acid (0.59 mg) and trehalose dihydrate (37 mg) and tromethamine for pH adjustment to 5.0.
- 0.605 mL containing 13.3 mg lonapegsomatropin-tcgd, succinic acid (0.71 mg) and trehalose dihydrate (44.8 mg) and tromethamine for pH adjustment to 5.0.
NONCLINICAL TOXICOLOGY SECTION
13 NONCLINICAL TOXICOLOGY
13.1 Carcinogenicity, Mutagenesis, Impairment of Fertility
Carcinogenicity studies have not been conducted with lonapegsomatropin-tcgd.
Lonapegsomatropin-tcgd was not mutagenic in the Ames test, in the human chromosomal aberration assay or in the rat bone marrow micronucleus test.
In an animal fertility study, lonapegsomatropin-tcgd was administered via subcutaneous injection to male and female rats before cohabitation, through mating to implantation.
Lonapegsomatropin-tcgd did not affect fertility or early embryo-fetal development at doses up to 20-fold the clinical pediatric dose of 0.24 mg/kg/week and approximately 46-fold the maximum clinical therapeutic dose for adult GHD of 6.3 mg hGH/week.
HOW SUPPLIED SECTION
16 HOW SUPPLIED/STORAGE AND HANDLING
How Supplied
SKYTROFA (lonapegsomatropin-tcgd) for injection is a sterile, preservative- free, white to off-white lyophilized powder available in a single-dose, dual- chamber, prefilled cartridge containing lonapegsomatropin-tcgd in one chamber and the diluent, Water for Injection, in the second chamber. The dual-chamber glass cartridge is available in 14 strengths (in somatropin equivalents) as described in Table 9.
Table 9: SKYTROFA Presentations|
SKYTROFA |
NDC |
|---|---|
|
0.7 mg |
73362-012-01 |
|
1.4 mg |
73362-013-01 |
|
1.8 mg |
73362-014-01 |
|
2.1 mg |
73362-015-01 |
|
2.5 mg |
73362-016-01 |
|
3 mg |
73362-003-01 |
|
3.6 mg |
73362-004-01 |
|
4.3 mg |
73362-005-01 |
|
5.2 mg |
73362-006-01 |
|
6.3 mg |
73362-007-01 |
|
7.6 mg |
73362-008-01 |
|
9.1 mg |
73362-009-01 |
|
11 mg |
73362-010-01 |
|
13.3 mg |
73362-011-01 |
Each carton contains 4 single-dose prefilled cartridges and 6 sterile, single- use, disposable 0.25 mm × 4 mm (31-gauge × 5/32 inch) needles. The cartridges are for use only with the SKYTROFA Auto-Injector, packaged in a separate carton. The SKYTROFA Auto-Injector is not supplied with SKYTROFA cartridges but is available for patients with a prescription for SKYTROFA through the Ascendis Pharma Customer Support by calling the toll-free number at 1-844-442-7236 (1-844-44ASCENDIS).
Storage and Handling
- For patients: Refrigerate SKYTROFA cartridges at 36°F to 46°F (2°C to 8°C) in the outer carton to protect from light until the expiration date. Do not freeze. Alternatively, SKYTROFA outer carton containing blistered cartridges may be stored at room temperature [up to 86°F (30°C)] for up to 6 months and can be returned to refrigeration within the 6 months. Write the date first removed from the refrigerator in the space provided on the outer carton. Do not use SKYTROFA beyond the expiration date or 6 months after the date it was first removed from refrigeration (whichever is earlier).
- For pharmacy long-term storage: Store SKYTROFA cartridges refrigerated at 36°F to 46°F (2°C to 8°C) in the outer carton to protect from light until the expiration date. Do not freeze.
SPL UNCLASSIFIED SECTION
© 2024 Ascendis Pharma. All rights reserved. SKYTROFA®, Ascendis®, TransCon®, the Ascendis Pharma logo and the company logo are trademarks owned by the Ascendis Pharma Group.
PATENT INFORMATION: www.ascendispharma.us/products/patents
Manufactured by:
Ascendis Pharma Endocrinology Division A/S
Tuborg Boulevard 12 Hellerup Denmark DK-2900
U.S. License Number 2165
For information about SKYTROFA contact:
Ascendis Pharma Endocrinology, Inc.
Princeton, New Jersey 08540, USA
1-844-442-7236 (1-844-44ASCENDIS)
www.Skytrofa.com
INSTRUCTIONS FOR USE SECTION
Quick Reference Guide
Skytrofa® Auto-Injector

For SKYTROFA cartridges
|
Single-patient use |
| ||
|
|


This is your
Quick Reference Guide
*Read your Instructions for Use for full instructions before using this Quick Reference Guide.
- For training video, go towww.skytrofa.com
- If you are unsure about your dose, contact your healthcare provider.
|
|
|
| ||
|
Instructions for Use |
Quick Reference Guide |
Training Video |


|
Ascendis Pharma Customer Support | |||
|
|
1-844-44ASCENDIS |

|
02 2025-07-18 8020100531_05 |
Parts overview

|
|
|
|

 Battery is fully
charged
Battery is fully
charged The SKYTROFA
The SKYTROFA
 At least 1
injection remaining, but charging recommended after use
At least 1
injection remaining, but charging recommended after use
 Battery needs
charging
Battery needs
chargingQuick troubleshooting
For all troubleshooting cases and details, see****Instructions for Use (IFU)
 IFU page 52
IFU page 52
Charging required (Step 2)
 IFU page 57
IFU page 57
|
|
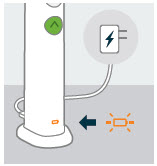
If the battery level is low: | |
|
|
You will see a flashing orange battery icon. | |
|
Do this: Insert charging cable into the SKYTROFA Auto-Injector at rear lower side and connect to a power outlet. |

SKYTROFA Auto-Injector not upright (Step 4 and 6.1)
 IFU page 54
IFU page 54
|
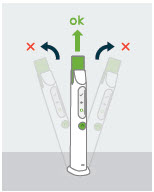
|
If the SKYTROFA Auto-Injector is not in an upright position during mixing and air removal: | |
|
|
You will hear a repeating notification sound. Mixing icon will flash slowly and progress bar will freeze. | |
|
Do this: Place the SKYTROFA Auto-Injector in an upright position and the mixing or air removal will continue. |
Skin contact lost (Step 9)
 IFU page 55
IFU page 55
|
|
If the green top is removed from skin before the injection is complete: | |
|
|
You will hear a repeating notification sound. Check mark icon will flash slowly and progress bar will freeze. | |
|
Do this: Press green top against skin and the injection will continue. |
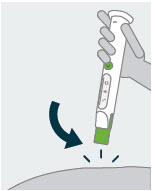

Prepare
1 Check and assemble cartridge and needle
|
1.1 |
1.2 |
1.3 | |
|
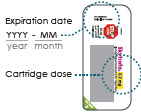
Check expiration date and cartridge dose on cartridge pack. If refrigerated, allow15 minutes to reach room temperature.
|
|
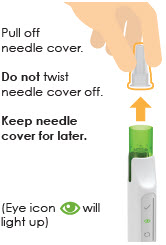
Screw needle straight and tightly on cartridge.Do not remove needle cover.
| |
|
Remove paper from needle. |
|



2 Turn on the auto-injector
|
|
| |
|
(You will hear2 loud beeps. Battery icon
|


 will light up
and green top will start flashing)
will light up
and green top will start flashing)3 Insert cartridge with attached needle
|
|
3.1 |
|
3.2 |
|
3.3 |



Mix
4 Wait while mixing
|
Wait 4 to 8 minutes for the auto-injector to mix medicine. |
|
|


If your auto-injector turns off automatically read Instructions for Use
 IFU page 60.
IFU page 60.
5 Turn the auto-injector up and down
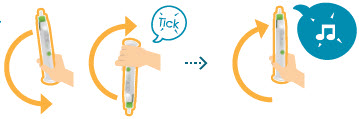
Turn the auto-injector up and down,listening for a tick sound each up and down cycle to make sure the turns are correct.Turn up and down correctly 5 to 10 times until you hear2 loud beeps and the whole progress bar, except the top element, lights up.Do not press the green button.
If you see a flashing orange mixing icon read
 IFU page 58.
IFU page 58.
6 Finish mixing
|
6.1 |
|
6.2 | ||
|
Keep the auto-injector upright on a flat surface until you hear2 loud beeps and the entire progress bar lights up. |
|

Inject
7 Check mixed medicine
8 Prepare for injection
|
8.1 |
8.2 |
8.3 | |
|
Choose an injection site: |
Make sure your hands are clean using soap and water or hand sanitizer. |
Clean injection site with alcohol wipe.Do not inject through clothes. |
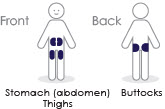


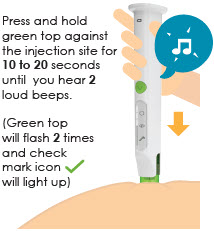
9 Inject medicine
|
9.1 |
9.2 | |
|
|
|

After injection
10 Remove cartridge
|
10.1 |
10.2 | |
|
|
|
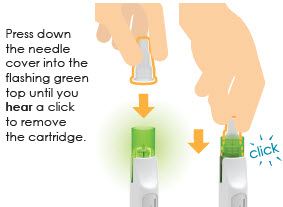

11 Check cartridge and throw away
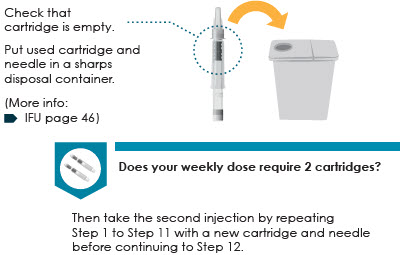
12 Store the auto-injector

USE IN SPECIFIC POPULATIONS SECTION
8 USE IN SPECIFIC POPULATIONS
8.1 Pregnancy
Risk Summary
There are no available data on lonapegsomatropin-tcgd use in pregnant patients to evaluate a drug-associated risk of major birth defects, miscarriage or adverse maternal or fetal outcomes. Available published data over several decades for somatropin, the active component of lonapegsomatropin-tcgd, have not identified a drug-associated risk of major birth defects, miscarriage or adverse maternal or fetal outcomes. In animal reproduction studies, there was no evidence of embryo-fetal or neonatal harm when pregnant rats were administered subcutaneous lonapegsomatropin-tcgd at doses up to 13-fold the clinical pediatric dose of 0.24 mg/kg/week and approximately 30-fold the maximum clinical therapeutic dose for adult GHD of 6.3 mg hGH/week (see Data).
The estimated background risk of birth defects and miscarriages for the indicated population is unknown. All pregnancies have a background risk of birth defect, loss, or other adverse outcomes. In the U.S. general population, the estimated background risk of major birth defects and miscarriages in clinically recognized pregnancies is 2% to 4% and 15% to 20%, respectively.
Data
Animal Data
No embryonic or fetal development toxicities occurred in rats administered subcutaneous lonapegsomatropin-tcgd at doses up to 13-fold the clinical pediatric dose of 0.24 mg/kg/week and approximately 30-fold the maximum clinical therapeutic dose for adult GHD of 6.3 mg hGH/week.
In a peri- and post-natal developmental study in rats, there were no adverse effects on the pregnant/lactating female or on development of the conceptus and the offspring following exposure of the female from implantation through weaning to doses of a structurally related pegylated somatropin prodrug up to 13-fold the clinical pediatric dose of 0.24 mg/kg/week and approximately 30-fold the maximum clinical therapeutic dose for adult GHD of 6.3 mg hGH/week.
8.2 Lactation
Risk Summary
There are no data on the presence of lonapegsomatropin-tcgd in human milk, effects on the breastfed infant, or effects on milk production. High molecular weight therapeutic proteins, including lonapegsomatropin-tcgd, are expected to have low passage into human milk and limited systemic exposure in the breastfed infant. Additionally, published data indicate that exogenous somatropin does not increase normal human milk concentrations of growth hormone. No adverse effects on the breastfed infant have been reported with somatropin. The developmental and health benefits of breastfeeding should be considered along with the mother's clinical need for SKYTROFA and any potential adverse effects on the breastfed infant from SKYTROFA or from the underlying maternal condition.
8.4 Pediatric Use
Safety and effectiveness of SKYTROFA have been established in pediatric patients 1 year and older and who weigh at least 11.5 kg. Pediatric use was established in a controlled study of 161 treatment-naïve pediatric patients ages 3 to 13 years and by supportive data in pediatric patients 1 year and older [see Adverse Reactions (6) and Clinical Studies (14)].
The safety and effectiveness of SKYTROFA in children less than 1 year of age have not been established.
Use of somatropin in pediatric patients with Prader-Willi syndrome has been associated with reports of sudden death. SKYTROFA is not indicated for the treatment of pediatric patients with growth failure due to genetically confirmed Prader-Willi syndrome [see Warnings and Precautions (5.13)].
8.5 Geriatric Use
Of the 249 patients who received SKYTROFA in clinical studies, 24 (10%) patients were 65 years of age and older, and 4 (2%) patients were 75 years of age and older. Geriatric patients may be at an increased risk for adverse reactions. Initiate SKYTROFA at 0.7 mg once weekly in patients 60 years of age and older, not on estrogen therapy [see Dosage and Administration (2.3)].
DRUG ABUSE AND DEPENDENCE SECTION
9 DRUG ABUSE AND DEPENDENCE
9.1 Controlled Substance
SKYTROFA is a prodrug of somatropin. Somatropin is not a controlled substance.
9.2 Abuse
Inappropriate use of somatropin may result in significant negative health consequences.
9.3 Dependence
Somatropin is not associated with drug related withdrawal adverse reactions.
CLINICAL STUDIES SECTION
14 CLINICAL STUDIES
14.1 Treatment-Naïve Pediatric Patients With Growth Hormone Deficiency
(NCT02781727)
A multi-center randomized, open-label, active-controlled, parallel-group phase 3 study was conducted in 161 treatment-naïve, prepubertal pediatric subjects with growth hormone deficiency (GHD); 105 subjects received once-weekly SKYTROFA, and 56 received daily somatropin. The dose in both arms was 0.24 mg/kg/week. The primary efficacy endpoint was annualized height velocity at Week 52.
The subjects ranged in age from 3.2 to 13.1 years with a mean of 8.5 years. One hundred thirty-two (82%) subjects were male and 29 (18%) were female. One subject was Asian, three were Black or African American, 152 were Caucasian, and five were categorized as "other." The subjects had a mean baseline height SDS (standard deviation score) of -2.9.
Treatment with once-weekly SKYTROFA for 52 weeks resulted in an annualized height velocity of 11.2 cm/year. Subjects treated with daily somatropin achieved an annualized height velocity of 10.3 cm/year after 52 weeks of treatment. Refer to Table 5.
Table 5: Annualized Height Velocity at Week 52 in Pediatric Treatment- Naïve Subjects with Growth Hormone Deficiency|
Once-Weekly SKYTROFA (N = 105) |
Daily Somatropin (N = 56) |
Estimate of Treatment Difference (95% CI) (SKYTROFA minus Daily Somatropin) | |
|---|---|---|---|
| |||
|
Annualized Height Velocity (cm/year)* |
11.2 |
10.3 |
0.9 |
Height SDS (change from baseline) was 1.1 in the SKYTROFA arm and 0.96 in the daily somatropin arm at Week 52. Refer to Table 6.
Table 6: Height SDS over 52 Weeks in Pediatric Treatment-Naïve Subjects with Growth Hormone Deficiency|
Once-Weekly SKYTROFA |
Daily Somatropin | |
|---|---|---|
|
Abbreviations: SDS, standard deviation score. | ||
| ||
|
Height SDS, baseline |
-2.9 |
-3.0 |
|
Height SDS, change from baseline* |
1.1 |
0.96 |
14.2 Adults With Growth Hormone Deficiency (NCT05171855)
A multi-center, randomized, parallel-group, placebo-controlled (double-blind), phase 3 study was conducted in 259 adults with GHD. Eighty-nine subjects received once-weekly SKYTROFA, 84 subjects received once-weekly placebo, and 86 subjects received open-label daily somatropin.
The mean (range) age at enrollment was 43 (23 to 81) years old, with 119 (46%) females (55 on oral estrogen) and 140 (54%) males, and the (SD) baseline body mass index of 28 (6.3) kg/m2. One subject was American Indian or Alaska Native, 1 was Black or African American, 28 were Asian, and 218 were Caucasian.
The primary efficacy endpoint, change in trunk percent (%) fat was measured by dual X-ray absorptiometry from baseline to Week 38 in the SKYTROFA group, compared to the placebo group (see Table 7).
Table 7: Change in Trunk Percent Fat from Baseline After 38 Weeks of Treatment in Adults with Growth Hormone Deficiency – SKYTROFA vs. Placebo|
Change from Baseline at Week 38 |
Once-Weekly SKYTROFA |
Once-Weekly Placebo |
LS Mean Difference [95% CI] |
P-value |
|---|---|---|---|---|
|
Abbreviations: LS, least squares; CI, confidence interval; kg, kilogram. | ||||
| ||||
|
Trunk percent fat (%)* |
-1.7 |
0.4 |
-2.0 [-2.9, -1.1] |
< 0.0001 |
Patients treated with daily somatropin achieved a change in trunk percent fat of -3.1% after 38 weeks. No formal statistical comparison between SKYTROFA and daily somatropin was conducted.
Change in total body lean mass and trunk fat mass from baseline after 38 weeks of treatment were secondary efficacy endpoints.
At 38 weeks, the change from baseline in total body lean mass was +1.6 kg for lonapegsomatropin and -0.1 kg for placebo (LS mean difference of 1.7 kg with 95% CI of 1.0 to 2.5, p-value < 0.0001).
At 38 weeks, the change from baseline in trunk fat mass was -0.5 kg for lonapegsomatropin and +0.2 kg for placebo (LS mean difference of -0.7 kg with 95% CI of -1.2 to -0.2, p-value = 0.005).
After 38 weeks, SKYTROFA treatment in adults with GHD resulted in normalization of IGF-1 SDS to 1.4 compared to -2.6 in placebo-treated patients. See Table 8.
Table 8: IGF-1 SDS at Baseline and after 38 Weeks of Treatment in Adults with Growth Hormone Deficiency – SKYTROFA vs. Placebo|
Time Point |
Once-Weekly SKYTROFA |
Once-Weekly Placebo |
|---|---|---|
|
Abbreviations: SD, standard deviation; SDS, standard deviation score. | ||
| ||
|
Baseline IGF-1 SDS, mean (SD) |
-2.6 (1.0) |
-2.7 (1.2) |
|
Week 38 IGF-1 SDS, mean (SD) * |
1.4 (1.9) |
-2.6 (1.3) |
The mean (SD) IGF-1 SDS level in daily somatropin treated patients was -2.82 (1) at baseline and 0.49 (1.98) at 38 weeks. No formal statistical comparison between SKYTROFA and daily somatropin was conducted.