
Paroxetine
These highlights do not include all the information needed to use safely and effectively. See full prescribing information for PAROXETINE TABLETS. PAROXETINE tablets, USP film coated for oral use Initial U.S. Approval: 1992
34909bfb-b4ad-25c4-e063-6294a90a0296
HUMAN PRESCRIPTION DRUG LABEL
Aug 13, 2025
NuCare Pharmaceuticals, Inc.
DUNS: 010632300
Products 1
Detailed information about drug products covered under this FDA approval, including NDC codes, dosage forms, ingredients, and administration routes.
paroxetine hydrochloride hemihydrate
Product Details
FDA regulatory identification and product classification information
FDA Identifiers
Product Classification
Product Specifications
INGREDIENTS (9)
Drug Labeling Information
PACKAGE LABEL.PRINCIPAL DISPLAY PANEL
PACKAGE/LABEL PRINCIPAL DISPLAY PANEL

CLINICAL PHARMACOLOGY SECTION
12 CLINICAL PHARMACOLOGY
12.1 Mechanism of Action
The mechanism of action of paroxetine tablets in the treatment of MDD, SAD, OCD, PD, GAD, and PTSD is unknown, but is presumed to be linked to potentiation of serotonergic activity in the central nervous system resulting from inhibition of neuronal reuptake of serotonin (5-hydroxy-tryptamine, 5-HT).
12.2 Pharmacodynamics
Studies at clinically relevant doses in humans have demonstrated that paroxetine blocks the uptake of serotonin into human platelets. In vitro studies in animals also suggest that paroxetine is a potent and highly selective inhibitor of neuronal serotonin reuptake (SSRI) and has only very weak effects on norepinephrine and dopamine neuronal reuptake.
12.3 Pharmacokinetics
Nonlinearity in pharmacokinetics is observed with increasing doses of paroxetine tablets.
In a meta-analysis of paroxetine from 4 studies done in healthy volunteers following multiple dosing of 20 mg/day to 40 mg/day, males did not exhibit a significantly lower C maxor AUC than females.
Absorption
Paroxetine hydrochloride is completely absorbed after oral dosing of a solution of the
hydrochloride salt. In a study in which normal male subjects (n = 15) received 30 mg tablets daily for 30 days, steady-state paroxetine concentrations were achieved by approximately 10 days for most subjects, although it may take substantially longer in an occasional patient. At steady state, mean values of C max, T max, C min, and T ½were 61.7 ng/mL (CV 45%), 5.2 hr. (CV 10%), 30.7 ng/mL (CV 67%), and 21 hours (CV 32%), respectively. The steady-state C maxand C minvalues were about 6 and 14 times what would be predicted from single-dose studies. Steady-state drug exposure based on AUC 0-24was about 8 times greater than would have been predicted from single-dose data in these subjects. The excess accumulation is a consequence of the fact that 1 of the enzymes that metabolizes paroxetine is readily saturable.
Effect of Food
The effects of food on the bioavailability of paroxetine were studied in subjects administered a single dose with and without food. AUC was only slightly increased (6%) when drug was administered with food but the C maxwas 29% greater, while the time to reach peak plasma concentration decreased from 6.4 hours post-dosing to 4.9 hours.
Distribution
Paroxetine distributes throughout the body, including the CNS, with only 1% remaining in the plasma.
Approximately 95% and 93% of paroxetine is bound to plasma protein at 100 ng/mL and 400 ng/mL, respectively. Under clinical conditions, paroxetine concentrations would normally be less than 400 ng/mL. Paroxetine does not alter the in vitroprotein binding of phenytoin or warfarin.
Elimination
Metabolism
The mean elimination half-life is approximately 21 hours (CV 32%) after oral dosing of 30 mg tablets daily for 30 days of paroxetine tablets.
In steady-state dose proportionality studies involving elderly and nonelderly patients, at doses of 20 mg to 40 mg daily for the elderly and 20 mg to 50 mg daily for the nonelderly, some nonlinearity was observed in both populations, again reflecting a saturable metabolic pathway. In comparison to C minvalues after 20 mg daily, values after 40 mg daily were only about 2 to 3 times greater than doubled.
Paroxetine is extensively metabolized after oral administration. The principal metabolites are polar and conjugated products of oxidation and methylation, which are readily cleared. Conjugates with glucuronic acid and sulfate predominate, and major metabolites have been isolated and identified. Data indicate that the metabolites have no more than 1/50 the potency of the parent compound at inhibiting serotonin uptake. The metabolism of paroxetine is accomplished in part by CYP2D6. Saturation of this enzyme at clinical doses appears to account for the nonlinearity of paroxetine kinetics with increasing dose and increasing duration of treatment. The role of this enzyme in paroxetine metabolism also suggests potential drug-drug interactions [see Drug Interactions ( 7)] . Pharmacokinetic behavior of paroxetine has not been evaluated in subjects who are deficient in CYP2D6 (poor metabolizers).
Excretion
Approximately 64% of a 30-mg oral solution dose of paroxetine was excreted in the urine with 2% as the parent compound and 62% as metabolites over a 10-day post-dosing period. About 36% was excreted in the feces (probably via the bile), mostly as metabolites and less than 1% as the parent compound over the 10-day post-dosing period.
Drug Interaction Studies
There are clinically significant, known drug interactions between paroxetine and other drugs [see Drug Interactions ( 7)].
Figure 1. Impact of Paroxetine on the Pharmacokinetics of Co-Administered Drugs (log scale)
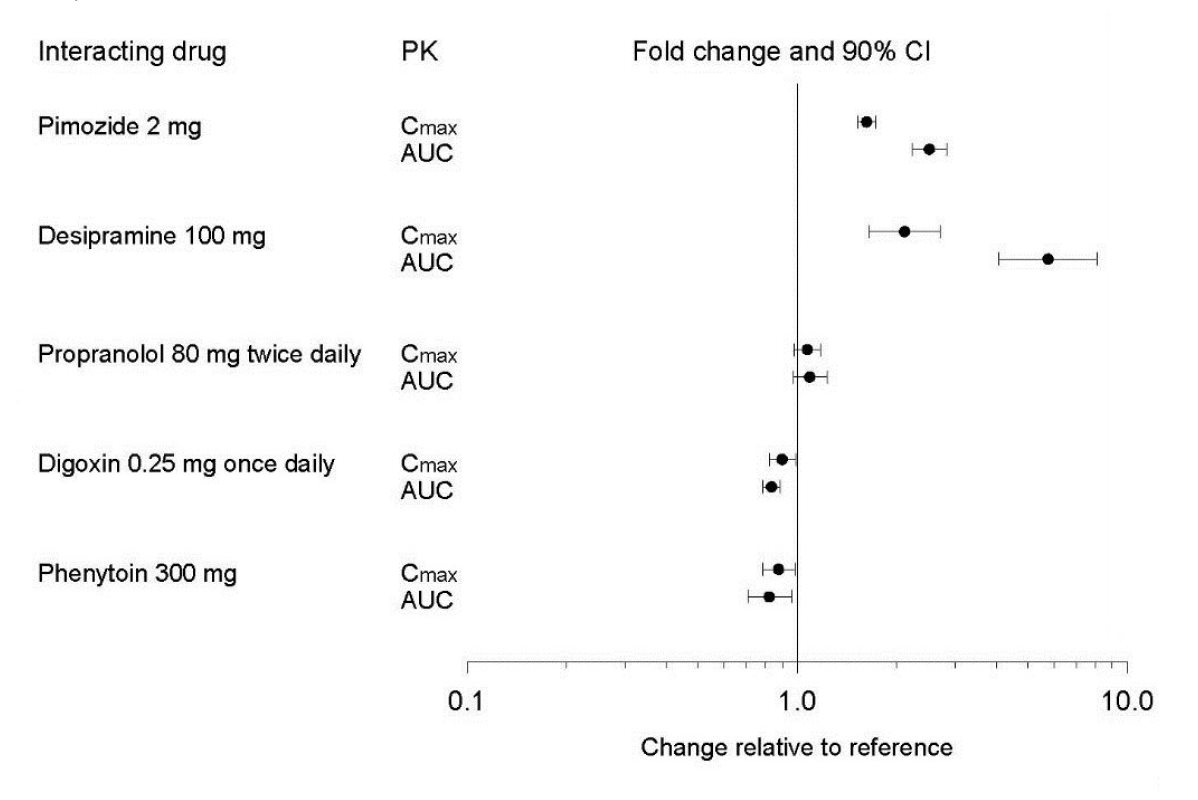
Figure 2. Impact of Co-Administered Drugs on the Pharmacokinetics of Paroxetine
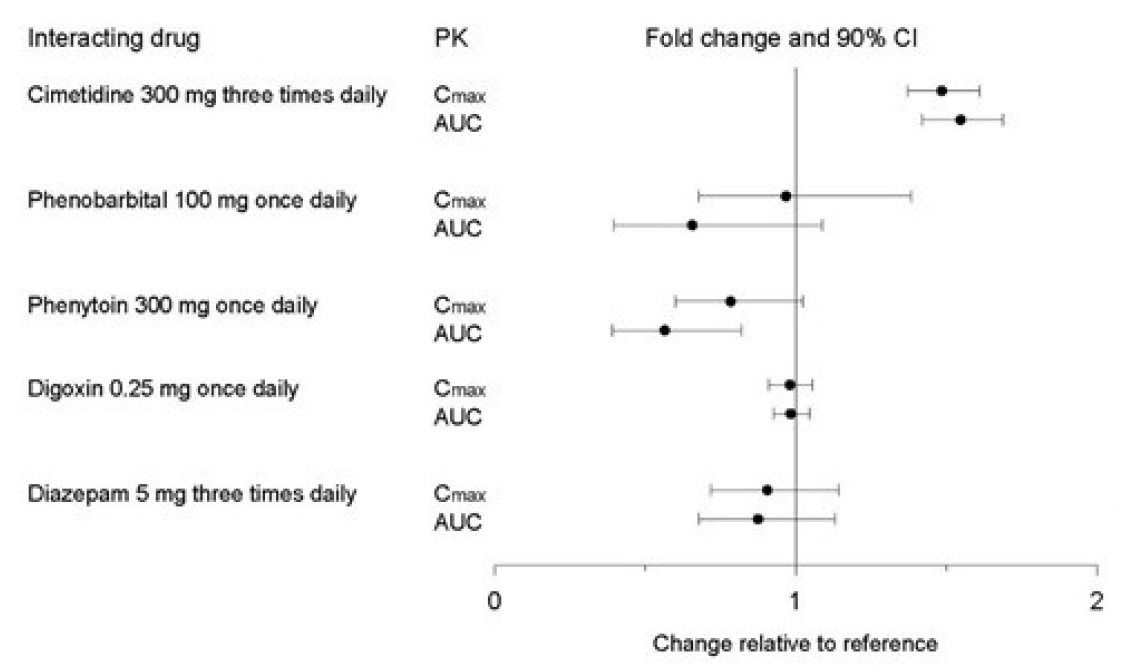
Theophylline:Reports of elevated theophylline levels associated with paroxetine tablets treatment have been reported. While this interaction has not been formally studied, it is recommended that theophylline levels be monitored when these drugs are concurrently administered.
Drugs Metabolized by Cytochrome CYP3A4
An in vivointeraction study involving the coadministration under steady-state conditions of paroxetine and terfenadine, a substrate for CYP3A4, revealed no effect of paroxetine on terfenadine pharmacokinetics. In addition, in vitrostudies have shown ketoconazole, a potent inhibitor of CYP3A4 activity, to be at least 100 times more potent than paroxetine as an inhibitor of the metabolism of several substrates for this enzyme, including terfenadine, triazolam, and cyclosporine. Paroxetine’s extent of inhibition of CYP3A4 activity is not expected to be of clinical significance.
Specific Populations
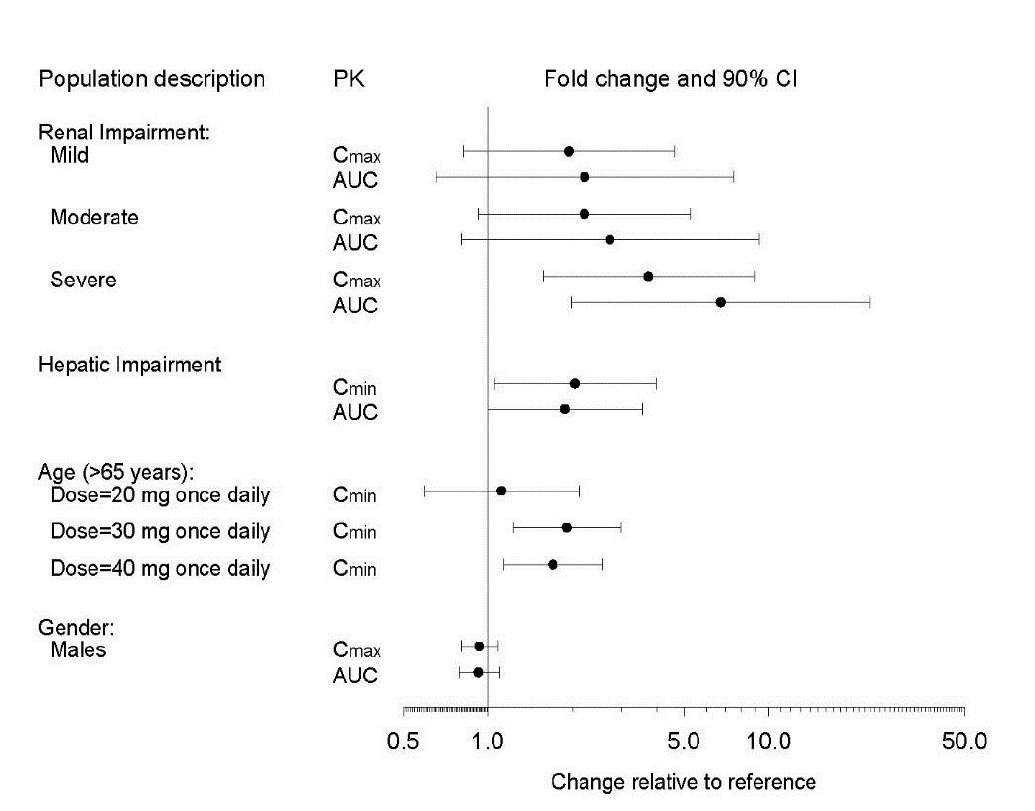
The impact of specific populations on the pharmacokinetics of paroxetine are shown in Figure 3.
The recommended starting dosage and maximum dosage of paroxetine tablets is reduced in elderly patients, patients with severe renal impairment, and patients with severe hepatic impairment [see Dosage and Administration ( 2.4)] .
Figure 3. Impact of Specific Population on the Pharmacokinetics of Paroxetine (log scale)
DESCRIPTION SECTION
11 DESCRIPTION
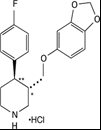
Paroxetine tablets contain paroxetine hydrochloride, an SSRI. It is the hydrochloride salt of a phenylpiperidine compound identified chemically as (-)- trans-4 R-(4'-fluorophenyl)-3 S-[(3',4'- methylenedioxyphenoxy) methyl] piperidine hydrochloride hemihydrate and has the empirical formula of C 19H 20FNO 3•HCl•1/2H 2O. The molecular weight is 374.8 (329.4 as free base). The structural formula of paroxetine hydrochloride is:
Paroxetine hydrochloride, USP is an odorless, off-white powder, having a melting point range of 120 oto 138 oC and a solubility of 5.4 mg/mL in water.
Paroxetine Tablets
Paroxetine tablets are for oral administration. Each film-coated tablet contains 10 mg, 20 mg, 30 mg, or 40 mg of paroxetine equivalent to 11.1 mg, 22.2 mg, 33.3 mg or 44.4 mg of paroxetine hydrochloride, respectively.
Inactive ingredients consist of glyceryl behenate, hypromellose, iron oxide red, iron oxide yellow, lactose monohydrate, magnesium stearate, polyethylene glycols and titanium dioxide.