
Leflunomide
These highlights do not include all the information needed to use LEFLUNOMIDE TABLETS safely and effectively. See full prescribing information for LEFLUNOMIDE TABLETS. LEFLUNOMIDE tablets, for oral use Initial U.S. Approval: 1998
e85c4eed-794c-409b-aed5-3c857b14b1fb
HUMAN PRESCRIPTION DRUG LABEL
Aug 11, 2023
Bryant Ranch Prepack
DUNS: 171714327
Products 1
Detailed information about drug products covered under this FDA approval, including NDC codes, dosage forms, ingredients, and administration routes.
Leflunomide
Product Details
FDA regulatory identification and product classification information
FDA Identifiers
Product Classification
Product Specifications
INGREDIENTS (12)
Drug Labeling Information
DRUG INTERACTIONS SECTION
7 DRUG INTERACTIONS
Following oral administration, leflunomide is metabolized to an active metabolite, teriflunomide, which is responsible for essentially all of leflunomide’s in vivo activity. Drug interaction studies have been conducted with both leflunomide and with its active metabolite, teriflunomide, where the metabolite was directly administered to the test subjects.
Effect of potent CYP and transporter inducers
Leflunomide is metabolized by CYP450 metabolizing enzymes. Concomitant use of leflunomide and rifampin, a potent inducer of CYP and transporters, increased the plasma concentration of teriflunomide by 40%. However, when co- administered with the metabolite, teriflunomide, rifampin did not affect its pharmacokinetics. No dosage adjustment is recommended for leflunomide when coadministered with rifampin. Because of the potential for leflunomide concentrations to continue to increase with multiple dosing, caution should be used if patients are to be receiving both leflunomide and rifampin [see Clinical Pharmacology (12.3)].
Effect on CYP2C8 substrates
Teriflunomide is an inhibitor of CYP2C8 in vivo. In patients taking leflunomide, exposure of drugs metabolized by CYP2C8 (e.g., paclitaxel, pioglitazone, repaglinide, rosiglitazone) may be increased. Monitor these patients and adjust the dose of the concomitant drug(s) metabolized by CYP2C8 as required [see Clinical Pharmacology (12.3)].
Effect on warfarin
Coadministration of leflunomide with warfarin requires close monitoring of the international normalized ratio (INR) because teriflunomide, the active metabolite of leflunomide, may decrease peak INR by approximately 25%.
Effect on oral contraceptives
Teriflunomide may increase the systemic exposures of ethinylestradiol and levonorgestrel. Consideration should be given to the type or dose of contraceptives used in combination with leflunomide [see Clinical Pharmacology (12.3)].
Effect on CYP1A2 substrates
Teriflunomide, the active metabolite of leflunomide, may be a weak inducer of CYP1A2 in vivo. In patients taking leflunomide, exposure of drugs metabolized by CYP1A2 (e.g., alosetron, duloxetine, theophylline, tizanidine) may be reduced. Monitor these patients and adjust the dose of the concomitant drug(s) metabolized by CYP1A2 as required [see Clinical Pharmacology (12.3)].
Effect on organic anion transporter 3 (OAT3) substrates
Teriflunomide inhibits the activity of OAT3 in vivo. In patients taking leflunomide, exposure of drugs which are OAT3 substrates (e.g., cefaclor, cimetidine, ciprofloxacin, penicillin G, ketoprofen, furosemide, methotrexate, zidovudine) may be increased. Monitor these patients and adjust the dose of the concomitant drug(s) which are OAT3 substrates as required [see Clinical Pharmacology (12.3)].
Effect on BCRP and organic anion transporting polypeptide B1 and B3 (OATP1B1/1B3) substrates
Teriflunomide inhibits the activity of BCRP and OATP1B1/1B3 in vivo. For a patient taking leflunomide, the dose of rosuvastatin should not exceed 10 mg once daily. For other substrates of BCRP (e.g., mitoxantrone) and drugs in the OATP family (e.g., methotrexate, rifampin), especially HMG-Co reductase inhibitors (e.g., atorvastatin, nateglinide, pravastatin, repaglinide, and simvastatin), consider reducing the dose of these drugs and monitor patients closely for signs and symptoms of increased exposures to the drugs while patients are taking leflunomide [see Clinical Pharmacology (12.3)].
- Drugs metabolized by CYP2C8 and OAT3 transporters: Monitor patients because teriflunomide may increase exposure of these drugs. (7)
- Teriflunomide may increase exposure of ethinylestradiol and levonorgestrel. Choose an appropriate oral contraceptive. (7)
- Drugs metabolized by CYP1A2: Monitor patients because teriflunomide may decrease exposure of these drugs. (7)
- Warfarin: Monitor INR as teriflunomide may decrease INR. (7)
- Drugs metabolized by BCRP and OATP1B1/B3 transporters: Monitor patients because teriflunomide may increase exposure of these drugs. (7)
- Rosuvastatin: The dose of rosuvastatin should not exceed 10 mg once daily in patients taking leflunomide. (7)
CLINICAL PHARMACOLOGY SECTION
12 CLINICAL PHARMACOLOGY
12.1 Mechanism of Action
Leflunomide is an isoxazole immunomodulatory agent that inhibits dihydroorotate dehydrogenase (a mitochondrial enzyme involved in de novo pyrimidine synthesis) and has antiproliferative activity. Several in vivo and in vitro experimental models have demonstrated an anti-inflammatory effect.
12.3 Pharmacokinetics
Following oral administration, leflunomide is metabolized to an active metabolite, teriflunomide, which is responsible for essentially all of leflunomide’s in vivo activity. Plasma concentrations of the parent drug, leflunomide, have been occasionally seen at very low concentrations. Studies of the pharmacokinetics of leflunomide have primarily examined the plasma concentrations of the active metabolite, teriflunomide.
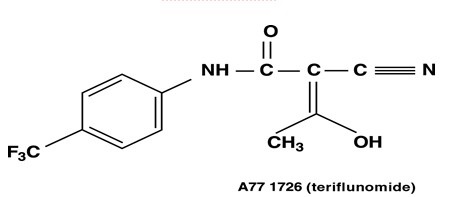
Absorption
Following oral administration, peak teriflunomide concentrations occurred between 6 to 12 hours after dosing. Due to the very long half-life of teriflunomide (18 to 19 days), a loading dose of 100 mg for 3 days was used in clinical studies to facilitate the rapid attainment of steady-state teriflunomide concentrations. Without a loading dose, it is estimated that attainment of steady-state plasma concentrations would require about two months of dosing. The resulting plasma concentrations following both loading doses and continued clinical dosing indicate that plasma teriflunomide concentrations are dose proportional.
Effect of Food
Co-administration of leflunomide tablets with a high fat meal did not have a significant impact on teriflunomide plasma concentrations.
Distribution
Teriflunomide is extensively bound to plasma protein (>99%) and is mainly distributed in plasma. The volume of distribution is 11 L after a single intravenous (IV) administration.
Elimination
Teriflunomide, the active metabolite of leflunomide, has a median half-life of 18 to 19 days in healthy volunteers. The elimination of teriflunomide can be accelerated by administration of cholestyramine or activated charcoal. Without use of an accelerated drug elimination procedure, it may take up to 2 years to reach plasma teriflunomide concentrations of less than 0.02 mg/L, due to individual variation in drug clearance [see Warnings and Precautions (5.3)]. After a single IV administration of the metabolite (teriflunomide), the total body clearance of teriflunomide was 30.5 mL/h.
Metabolism
In vitro inhibition studies in human liver microsomes suggest that cytochrome P450 (CYP) 1A2, 2C19 and 3A4 are involved in leflunomide metabolism. In vivo, leflunomide is metabolized to one primary (teriflunomide) and many minor metabolites. In vitro, teriflunomide is not metabolized by CYP450 or flavin monoamine oxidase enzymes. The parent compound is rarely detectable in plasma.
Excretion
Teriflunomide, the active metabolite of leflunomide, is eliminated by direct biliary excretion of unchanged drug as well as renal excretion of metabolites. Over 21 days, 60.1% of the administered dose is excreted via feces (37.5%) and urine (22.6%). After an accelerated elimination procedure with cholestyramine, an additional 23.1% was recovered (mostly in feces).
Studies with both hemodialysis and CAPD (chronic ambulatory peritoneal dialysis) indicate that teriflunomide is not dialyzable.
Specific Populations
Gender. Gender has not been shown to cause a consistent change in the in vivo pharmacokinetics of teriflunomide.
Smoking. A population based pharmacokinetic analysis of the clinical trial data indicates that smokers have a 38% increase in clearance over non-smokers; however, no difference in clinical efficacy was seen between smokers and nonsmokers.
Drug Interaction Studies
Drug interaction studies have been conducted with both leflunomide and with its active metabolite, teriflunomide, where the metabolite was directly administered to the test subjects.
The Potential Effect of Other Drugs on Leflunomide
- Potent CYP and transporter inducers:
Following concomitant administration of a single dose of leflunomide to subjects receiving multiple doses of rifampin, teriflunomide peak concentrations were increased (~40%) over those seen when leflunomide was given alone [see Drug Interactions (7)].
- An in vivo interaction study with leflunomide and cimetidine (non-specific weak CYP inhibitor) has demonstrated a lack of a significant impact on teriflunomide exposure.
The Potential Effect of Leflunomide on Other Drugs
- CYP2C8 Substrates
There was an increase in mean repaglinide Cmaxand AUC (1.7-and 2.4-fold, respectively), following repeated doses of teriflunomide and a single dose of 0.25 mg repaglinide, suggesting that teriflunomide is an inhibitor of CYP2C8 in vivo. The magnitude of interaction could be higher at the recommended repaglinide dose [see Drug Interactions (7)].
- CYP1A2 Substrates
Repeated doses of teriflunomide decreased mean Cmax and AUC of caffeine by 18% and 55%, respectively, suggesting that teriflunomide may be a weak inducer of CYP1A2 in vivo.
- OAT3 Substrates
There was an increase in mean cefaclor Cmax and AUC (1.43-and 1.54-fold, respectively), following repeated doses of teriflunomide, suggesting that teriflunomide is an inhibitor of organic anion transporter 3 (OAT3) in vivo[see Drug Interactions (7)].
- BCRP and OATP1B1/1B3 Substrates
There was an increase in mean rosuvastatin Cmax and AUC (2.65-and 2.51-fold, respectively), following repeated doses of teriflunomide, suggesting that teriflunomide is an inhibitor of BCRP transporter and organic anion transporting polypeptide 1B1 and 1B3 (OATP1B1/1B3) [see Drug Interactions (7)].
- Oral Contraceptives
There was an increase in mean ethinylestradiol Cmax and AUC0 to 24 (1.58-and 1.54-fold, respectively) and levonorgestrel Cmax and AUC0 to 24 (1.33-and 1.41-fold, respectively) following repeated doses of teriflunomide [see Drug Interactions (7)].
- Teriflunomide did not affect the pharmacokinetics of bupropion (a CYP2B6 substrate), midazolam (a CYP3A4 substrate), S-warfarin (a CYP2C9 substrate), omeprazole (a CYP2C19 substrate), and metoprolol (a CYP2D6 substrate).
OVERDOSAGE SECTION
10 OVERDOSAGE
There have been reports of chronic overdose in patients taking leflunomide at daily dose up to five times the recommended daily dose and reports of acute overdose in adults and children. Adverse events were consistent with the safety profile for leflunomide [See Adverse Reactions (6)]. The most frequent adverse events observed were diarrhea, abdominal pain, leukopenia, anemia and elevated liver function tests.
In the event of a significant overdose or toxicity, perform an accelerated drug elimination procedure to accelerate elimination [see Warnings and Precautions (5.3)].
Studies with both hemodialysis and CAPD (chronic ambulatory peritoneal dialysis) indicate that teriflunomide, the primary metabolite of leflunomide, is not dialyzable [See Clinical Pharmacology (12.3)].
NONCLINICAL TOXICOLOGY SECTION
13 NONCLINICAL TOXICOLOGY
13.1 Carcinogenesis, Mutagenesis, Impairment of Fertility
No evidence of carcinogenicity was observed in a 2-year bioassay in rats at oral doses of leflunomide up to the maximally tolerated dose of 6 mg/kg (approximately 1/40 the maximum human teriflunomide systemic exposure based on AUC). However, male mice in a 2-year bioassay exhibited an increased incidence in lymphoma at an oral dose of 15 mg/kg, the highest dose studied (1.7 times the human teriflunomide exposure based on AUC). Female mice, in the same study, exhibited a dose-related increased incidence of bronchoalveolar adenomas and carcinomas combined beginning at 1.5 mg/kg (approximately 1/10 the human teriflunomide exposure based on AUC). The significance of the findings in mice relative to the clinical use of leflunomide is not known.
Leflunomide was not mutagenic in the Ames assay, the unscheduled DNA synthesis assay, or in the HGPRT gene mutation assay. In addition, leflunomide was not clastogenic in the in vivo mouse micronucleus assay or in the in vivo Chinese hamster bone marrow cell cytogenic test. However, 4-trifluoromethylaniline (TFMA), a minor metabolite of leflunomide, was mutagenic in the Ames assay and in the HGPRT gene mutation assay, and was clastogenic in the in vitro Chinese hamster cell chromosomal aberration assay. TFMA was not clastogenic in the in vivo mouse micronucleus assay or in the in vivo Chinese hamster bone marrow cell cytogenic test.
Leflunomide had no effect on fertility or reproductive performance in either male or female rats at oral doses up to 4.0 mg/kg (approximately 1/30 the human teriflunomide exposure based on AUC) [see Use in Specific Populations (8.1, 8.6)].