
Lenalidomide
These highlights do not include all the information needed to use LENALIDOMIDE CAPSULES safely and effectively. See full prescribing information for LENALIDOMIDE CAPSULES. LENALIDOMIDE capsules, for oral use Initial U.S. Approval: 2005
9943e098-20b0-dbb3-af12-7816eedd3f70
HUMAN PRESCRIPTION DRUG LABEL
Sep 19, 2025
Apotex Corp.
DUNS: 845263701
Products 6
Detailed information about drug products covered under this FDA approval, including NDC codes, dosage forms, ingredients, and administration routes.
Lenalidomide
Product Details
FDA regulatory identification and product classification information
FDA Identifiers
Product Classification
Product Specifications
INGREDIENTS (11)
Lenalidomide
Product Details
FDA regulatory identification and product classification information
FDA Identifiers
Product Classification
Product Specifications
INGREDIENTS (11)
Lenalidomide
Product Details
FDA regulatory identification and product classification information
FDA Identifiers
Product Classification
Product Specifications
INGREDIENTS (12)
Lenalidomide
Product Details
FDA regulatory identification and product classification information
FDA Identifiers
Product Classification
Product Specifications
INGREDIENTS (11)
Lenalidomide
Product Details
FDA regulatory identification and product classification information
FDA Identifiers
Product Classification
Product Specifications
INGREDIENTS (11)
Lenalidomide
Product Details
FDA regulatory identification and product classification information
FDA Identifiers
Product Classification
Product Specifications
INGREDIENTS (13)
Drug Labeling Information
PACKAGE LABEL.PRINCIPAL DISPLAY PANEL
PRINCIPAL DISPLAY PANEL 25 mg
Representative sample of labeling (see HOW SUPPLIED section for complete listing):
APOTEX CORP. NDC 60505-4537-2
Lenalidomide Capsules
25 mg
Rx Only
21 Capsules

DESCRIPTION SECTION
11 DESCRIPTION
Lenalidomide, a thalidomide analogue, is an immunomodulatory agent with antiangiogenic and antineoplastic properties. The chemical name is 3-(4-amino-1-oxo 1,3- dihydro-2H-isoindol-2-yl) piperidine-2,6-dione and it has the following chemical structure:
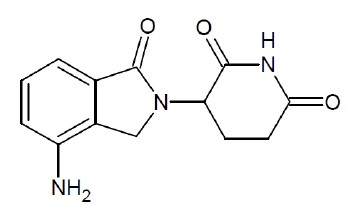
3-(4-amino-1-oxo 1,3-dihydro-2H-isoindol-2-yl) piperidine-2,6-dione
The molecular formula for lenalidomide is C13H13N3O3, and the gram molecular weight is 259.26 g/mol. Lenalidomide is an off-white to pale-yellow powder. It is freely soluble in dimethylsulfoxide, slightly soluble in acetonitrile and methanol, very slightly soluble in ethyl acetate, and practically insoluble, or insoluble in water, absolute ethanol, hexane and isopropanol. Lenalidomide has an asymmetric carbon atom and can exist as the optically active forms S(-) and R(+), and is produced as a racemic mixture with a net optical rotation of zero.
Lenalidomide is available in 2.5 mg, 5 mg, 10 mg, 15 mg 20 mg and 25 mg capsules for oral administration. Each capsule contains crystalline anhydrous lenalidomide as the active ingredient and the following inactive ingredients: anhydrous lactose, croscarmellose sodium, magnesium stearate and microcrystalline cellulose. The 5 mg and 25 mg capsule shell contains gelatin, titanium dioxide and black ink. The 2.5 mg, 10 mg and 20 mg capsule shell contains FD&C blue #2, gelatin, iron oxide yellow, titanium dioxide and black ink. The 15 mg capsule shell contains FD&C blue #2, gelatin, titanium dioxide and black ink. The capsule imprinting black ink contains ammonium hydroxide 28%, iron oxide black, propylene glycol and shellac.
CLINICAL STUDIES SECTION
14 CLINICAL STUDIES
14.1 Multiple Myeloma
Randomized, Open-Label Clinical Trial in Patients with Newly Diagnosed MM:
A randomized multicenter, open-label, 3-arm trial of 1,623 patients, was conducted to compare the efficacy and safety of lenalidomide capsules and low- dose dexamethasone (Rd) given for 2 different durations of time to that of melphalan, prednisone and thalidomide (MPT) in newly diagnosed MM patients who were not a candidate for stem cell transplant. In the first arm of the study, Rd was given continuously until progressive disease [Arm Rd Continuous]. In the second arm, Rd was given for up to eighteen 28-day cycles [72 weeks, Arm Rd18]). In the third arm, melphalan, prednisone and thalidomide (MPT) was given for a maximum of twelve 42-day cycles (72 weeks). For the purposes of this study, a patient who was < 65 years of age was not a candidate for SCT if the patient refused to undergo SCT therapy or the patient did not have access to SCT due to cost or other reasons. Patients were stratified at randomization by age (≤75 versus >75 years), stage (ISS Stages I and II versus Stage III), and country.
Patients in the Rd Continuous and Rd18 arms received lenalidomide capsules 25 mg once daily on Days 1 to 21 of 28-day cycles. Dexamethasone was dosed 40 mg once daily on Days 1, 8, 15, and 22 of each 28-day cycle. For patients over > 75 years old, the starting dose of dexamethasone was 20 mg orally once daily on days 1,8,15, and 22 of repeated 28-day cycles. Initial dose and regimens for Rd Continuous and Rd18 were adjusted according to age and renal function. All patients received prophylactic anticoagulation with the most commonly used being aspirin.
The demographics and disease-related baseline characteristics of the patients were balanced among the 3 arms. In general, study subjects had advanced-stage disease. Of the total study population, the median age was 73 in the 3 arms with 35% of total patients > 75 years of age; 59% had ISS Stage I/II; 41% had ISS stage III; 9% had severe renal impairment (creatinine clearance [CLcr] < 30 mL/min); 23% had moderate renal impairment (CLcr > 30 to 50 mL/min; 44% had mild renal impairment (CLcr > 50 to 80 mL/min). For ECOG Performance Status, 29% were Grade 0, 49% Grade 1, 21% Grade 2, 0.4% ≥ Grade 3.
The primary efficacy endpoint, progression-free survival (PFS), was defined as the time from randomization to the first documentation of disease progression as determined by Independent Response Adjudication Committee (IRAC), based on International Myeloma Working Group [IMWG] criteria or death due to any cause, whichever occurred first during the study until the end of the PFS follow-up phase. For the efficacy analysis of all endpoints, the primary comparison was between Rd Continuous and MPT arms. The efficacy results are summarized in the table below. PFS was significantly longer with Rd Continuous than MPT: HR 0.72 (95% CI: 0.61-0.85 p <0.0001). A lower percentage of subjects in the Rd Continuous arm compared with the MPT arm had PFS events (52% versus 61%, respectively). The improvement in median PFS time in the Rd Continuous arm compared with the MPT arm was 4.3 months. The myeloma response rate was higher with Rd Continuous compared with MPT (75.1% versus 62.3%); with a complete response in 15.1% of Rd Continuous arm patients versus 9.3% in the MPT arm. The median time to first response was 1.8 months in the Rd Continuous arm versus 2.8 months in the MPT arm.
For the interim OS analysis with 03 March 2014 data cutoff, the median follow- up time for all surviving patients is 45.5 months, with 697 death events, representing 78% of prespecified events required for the planned final OS analysis (697/896 of the final OS events). The observed OS HR was 0.75 for Rd Continuous versus MPT (95% CI = 0.62, 0.90).
Table 13: Overview of Efficacy Results – Study MM-020 (Intent-to-treat Population)
|
Rd Continuous |
Rd18 |
MPT**** | |
|
**PFS – IRAC (months)**g | |||
|
Number of PFS events |
278 (52) |
348 (64.3) |
334 (61.1) |
|
Mediana PFS time, months (95% CI)b |
25.5 (20.7, 29.4) |
20.7 (19.4, 22) |
21.2 (19.3, 23.2) |
|
HR [95% CI]c; p-valued | |||
|
Rd Continuous vs MPT |
0.72 (0.61, 0.85); | ||
|
Rd Continuous vs Rd18 |
0.70 (0.60, 0.82) | ||
|
Rd18 vs MPT |
1.03 (0.89, 1.20) | ||
|
**Overall Survival (months)**h | |||
|
Number of Death events |
208 (38.9) |
228 (42.1) |
261 (47.7) |
|
Mediana OS time, months (95% CI)b |
58.9 (56, NE)f |
56.7 (50.1, NE) |
48.5 (44.2, 52 ) |
|
HR [95% CI]c | |||
|
Rd Continuous vs MPT |
0.75 (0.62, 0.90) | ||
|
Rd Continuous vs Rd18 |
0.91 (0.75, 1.09) | ||
|
Rd18 vs MPT |
0.83 (0.69, 0.99) | ||
|
Response Ratee**– IRAC, n (%)**g | |||
|
CR |
81 (15.1) |
77 (14.2) |
51 (9.3) |
|
VGPR |
152 (28.4) |
154 (28.5) |
103 (18.8) |
|
PR |
169 (31.6) |
166 (30.7) |
187 (34.2) |
|
Overall response: CR, VGPR, or PR |
402 (75.1) |
397 (73.4) |
341 (62.3) |
CR = complete response; d = low-dose dexamethasone; HR = hazard ratio; IRAC = Independent Response Adjudication Committee; M = melphalan; NE = not estimable; OS = overall survival; P = prednisone; PFS = progression-free survival; PR = partial response; R = lenalidomide; Rd Continuous = Rd given until documentation of progressive disease; Rd18 = Rd given for ≤ 18 cycles; T = thalidomide; VGPR = very good partial response; vs = versus.
a The median is based on the Kaplan-Meier estimate.
b The 95% Confidence Interval (CI) about the median.
c Based on Cox proportional hazards model comparing the hazard functions associated with the indicated treatment arms.
d The p-value is based on the unstratified log-rank test of Kaplan-Meier curve differences between the indicated treatment arms.
e Best assessment of response during the treatment phase of the study.
f Including patients with no response assessment data or whose only assessment was “response not evaluable.”
g Data cutoff date = 24 May 2013.
h Data cutoff date = 3 March 2014.
Kaplan-Meier Curves of Progression-free Survival Based on IRAC Assessment (ITT MM Population) Between Arms Rd Continuous, Rd18 and MPT Cutoff date: 24 May 2013
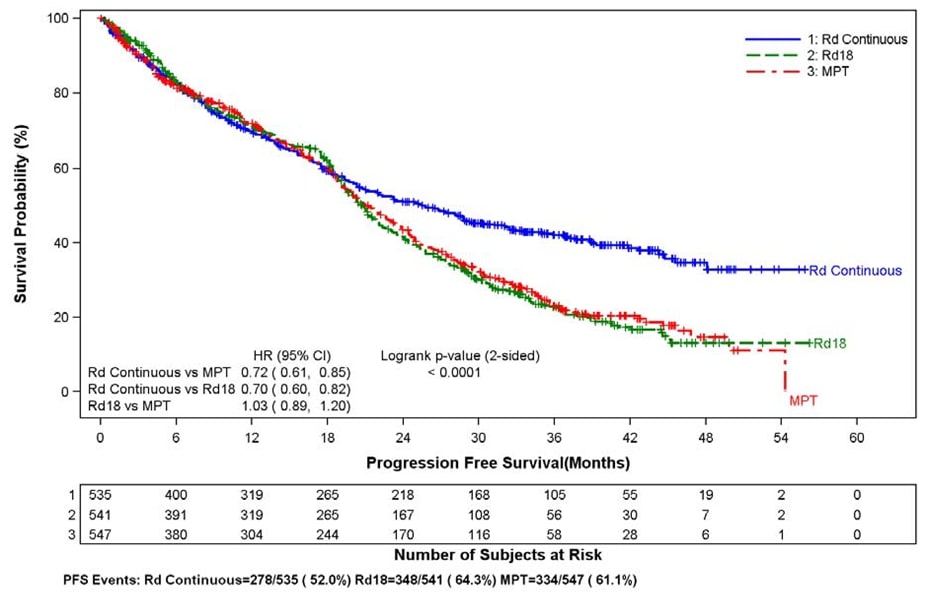
CI = confidence interval; d = low-dose dexamethasone; HR = hazard ratio; IRAC = Independent Response Adjudication Committee; M = melphalan; P = prednisone; R = lenalidomide; Rd Continuous = Rd given until documentation of progressive disease; Rd18 = Rd given for ≤ 18 cycles; T = thalidomide.
Kaplan-Meier Curves of Overall Survival (ITT MM Population) Between Arms Rd Continuous, Rd18 and MPT Cutoff date: 03 Mar 2014
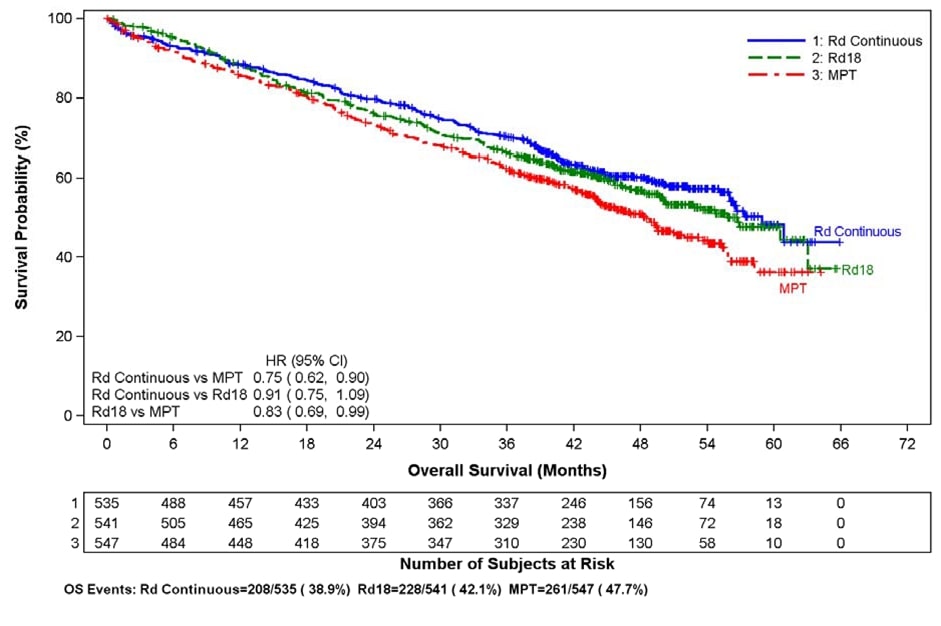
CI = confidence interval; d = low-dose dexamethasone; HR = hazard ratio; M = melphalan; P = prednisone; R = lenalidomide; Rd Continuous = Rd given until documentation of progressive disease; Rd18 = Rd given for ≤18 cycles; T = thalidomide.
Randomized, Placebo-Controlled Clinical Trials - Maintenance Following Auto- HSCT:
Two multicenter, randomized, double-blind, parallel group, placebo-controlled studies were conducted to evaluate the efficacy and safety of lenalidomide maintenance therapy in the treatment of MM patients after auto-HSCT. In Maintenance Study 1, patients between 18 and 70 years of age who had undergone induction therapy followed by auto-HSCT were eligible. Induction therapy must have occurred within 12 months. Within 90-100 days after auto-HSCT, patients with at least a stable disease response were randomized 1:1 to receive either lenalidomide or placebo maintenance. In Maintenance Study 2, patients aged < 65 years at diagnosis who had undergone induction therapy followed by auto- HSCT and had achieved at least a stable disease response at the time of hematologic recovery were eligible. Within 6 months after auto-HSCT, patients were randomized 1:1 to receive either lenalidomide or placebo maintenance. Patients eligible for both trials had to have CLcr ≥30 mL/minute.
In both studies, the lenalidomide maintenance dose was 10 mg once daily on days 1-28 of repeated 28-day cycles, could be increased to 15 mg once daily after 3 months in the absence of dose-limiting toxicity, and treatment was to be continued until disease progression or patient withdrawal for another reason. The dose was reduced, or treatment was temporarily interrupted or stopped, as needed to manage toxicity. A dose increase to 15 mg once daily occurred in 135 patients (58%) in Maintenance Study 1, and in 185 patients (60%) in Maintenance Study 2.
The demographics and disease-related baseline characteristics of the patients were similar across the two studies and reflected a typical MM population after auto- HSCT (see Table 14).
Table 14: Baseline Demographic and Disease-Related Characteristics – MM Maintenance Studies 1 and 2
|
Maintenance Study 1 |
Maintenance Study 2 | |||
|
Lenalidomide****Capsules N = 231 |
Placebo N = 229 |
Lenalidomide****Capsules N = 307 |
Placebo N = 307 | |
|
Age (years) | ||||
|
Median |
58 |
58 |
57.5 |
58.1 |
|
(Min, max) |
(29, 71) |
(39, 71) |
(22.7, 68.3) |
(32.3, 67) |
|
Sex, n (%) | ||||
|
Male |
121 (52) |
129 (56) |
169 (55) |
181 (59) |
|
Female |
110 (48) |
100 (44) |
138 (45) |
126 (41) |
|
ISS Stage at Diagnosis, n (%) | ||||
|
Stage I or II |
120 (52) |
131 (57) |
232 (76) |
250 (81) |
|
Stage I |
62 (27) |
85 (37) |
128 (42) |
143 (47) |
|
Stage II |
58 (25) |
46 (20) |
104 (34) |
107 (35) |
|
Stage III |
39 (17) |
35 (15) |
66 (21) |
46 (15) |
|
Missing |
72 (31) |
63 (28) |
9 (3) |
11 (4) |
|
CrCl at Post-auto-HSCT, n (%) | ||||
|
< 50 mL/min |
23 (10) |
16 (7) |
10 (3) |
9 (3) |
|
≥ 50 mL/min |
201 (87) |
204 (89) |
178 (58) |
200 (65) |
|
Missing |
7 (3) |
9 (4) |
119 (39) |
98 (32) |
Data cutoff date = 1 March 2015.
The major efficacy endpoint of both studies was PFS defined from randomization to the date of progression or death, whichever occurred first; the individual studies were not powered for an overall survival endpoint. Both studies were unblinded upon the recommendations of their respective data monitoring committees and after surpassing the respective thresholds for preplanned interim analyses of PFS. After unblinding, patients continued to be followed as before. Patients in the placebo arm of Maintenance Study 1 were allowed to cross over to receive lenalidomide capsules before disease progression (76 patients [33%] crossed over to lenalidomide capsules); patients in Maintenance Study 2 were not recommended to cross over. The efficacy results are summarized in the following table. In both studies, the primary analysis of PFS at unblinding was significantly longer with lenalidomide capsules compared to placebo: Maintenance Study 1 HR 0.38 (95% CI: 0.27-0.54 p <0.001) and Maintenance Study 2 HR 0.50 (95% CI: 0.39-0.64 p <0.001). For both studies, PFS was updated with a cutoff date of 1 March 2015 as shown in the table and the following Kaplan Meier graphs. With longer follow-up (median 72.4 and 86.0 months, respectively), the updated PFS analyses for both studies continue to show a PFS advantage for lenalidomide capsules compared to placebo: Maintenance Study 1 HR 0.38 (95% CI: 0.28-0.50) with median PFS of 68.6 months and Maintenance Study 2 HR 0.53 (95% CI: 0.44-0.64) with median PFS of 46.3 months.
Descriptive analysis of OS data with a cutoff date of 1 February 2016 are provided in Table 15. Median follow-up time was 81.6 and 96.7 months for Maintenance Study 1 and Maintenance Study 2, respectively. Median OS was 111.0 and 84.2 months for lenalidomide capsules and placebo, respectively, for Maintenance Study 1, and 105.9 and 88.1 months, for lenalidomide capsules and placebo, respectively, for Maintenance Study 2.
Table 15: Progression-free Survival and Overall Survival from Randomization in MM Maintenance Studies 1 and 2 (ITT Post-Auto-HSCT Population)
|
Maintenance Study 1 |
Maintenance Study 2 | |||
|
Lenalidomide Capsules**** |
Placebo |
Lenalidomide Capsules**** |
Placebo**** | |
|
PFS at Unblinding | ||||
|
PFS Events n (%) |
**** 46 (20) |
**** 98 (43) |
**** 103 (34) |
**** 160 (52) |
|
**** Median in months [95% CI] |
33.9 [NE, NE] |
19 [16.2, 25.6] |
41.2 [38.3, NE] |
23.0 [21.2, 28.0] |
|
Hazard Ratio [95% CI] |
0.38 [0.27, 0.54] |
0.50 [0.39, 0.64] | ||
|
Log-rank Test p-value |
**** <0.001 |
**** <0.001 | ||
|
PFS at Updated Analysis****1 March 2015 (Studies 1 and 2) | ||||
|
**** PFS Events n (%) |
**** 97 (42) |
**** 116 (51) |
**** 191 (62) |
**** 248 (81) |
|
**** Median in months [95% CI] |
68.6 [52.8, NE] |
22.5 [18.8, 30.0] |
46.3 [40.1, 56.6] |
23.8 [21.0, 27.3] |
|
Hazard Ratio [95% CI] |
0.38 [0.28, 0.50] |
0.53 [0.44, 0.64] | ||
|
OS at Updated Analysis****1 Feb 2016 (Studies 1 and 2) | ||||
|
**** OS Events n (%) |
**** 82 (35) |
**** 114 (50) |
**** 143 (47) |
**** 160 (52) |
|
**** Median in months [95% CI] |
111 [101.8, NE] |
84.2 [71.0, 102.7] |
105.9 [88.8, NE] |
88.1 [80.7, 108.4] |
|
Hazard Ratio [95% CI] |
0.59 [0.44, 0.78] |
0.90 [0.72, 1.13] |
Date of Unblinding in Maintenance Study 1 and 2 = 17 December 2009 and 7 July 2010, respectively.
Auto-HSCT = autologous hematopoietic stem cell transplantation; CI = confidence interval; ITT = intent to treat; NE = not estimable;
PFS = progression-free survival.
PFS at time of unblinding for Maintenance Study 2 was based on assessment by an Independent Review Committee. All other PFS analyses were based on assessment by investigator.
Note: The median is based on Kaplan-Meier estimate, with 95% CIs about the median overall PFS time. Hazard ratio is based on a proportional hazards model stratified by stratification factors comparing the hazard functions associated with treatment arms (lenalidomide capsules: placebo).
Kaplan-Meier Curves of Progression-free Survival from Randomization**(ITT Post-Auto-HSCT Population) in MM Maintenance Study 1 between Lenalidomide Capsules and Placebo Arms****(Updated Cutoff Date 1 March 2015)**
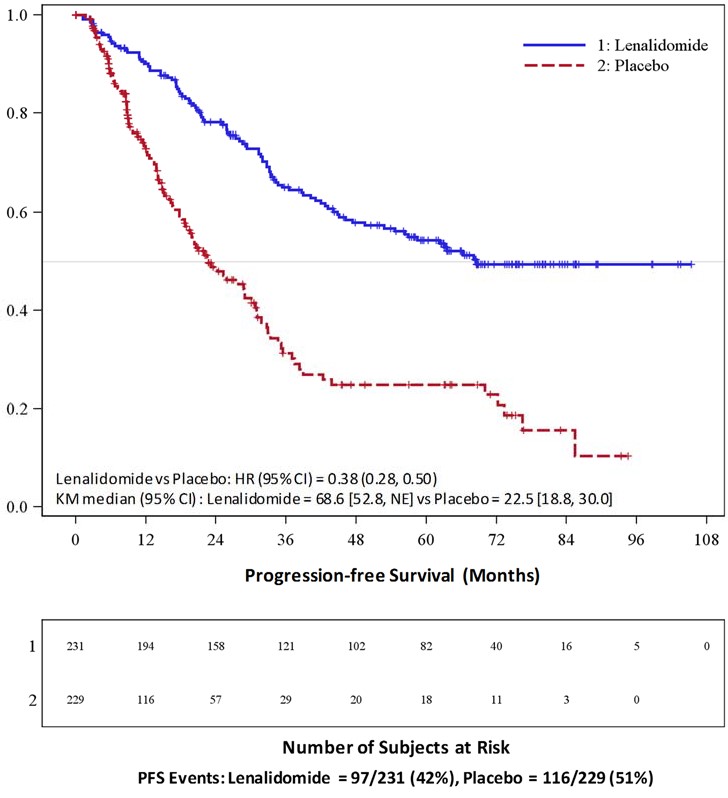 Auto-HSCT =
autologous hematopoietic stem cell transplantation; CI = confidence interval;
HR = hazard ratio; ITT = intent to treat; KM = Kaplan-Meier; PFS =
progression-free survival; vs = versus.
Auto-HSCT =
autologous hematopoietic stem cell transplantation; CI = confidence interval;
HR = hazard ratio; ITT = intent to treat; KM = Kaplan-Meier; PFS =
progression-free survival; vs = versus.
Kaplan-Meier Curves of Progression-free Survival from Randomization (ITT Post-Auto-HSCT Population) in MM Maintenance Study 2 between Lenalidomide Capsules and Placebo Arms**(Updated Cutoff Date 1 March 2015)**
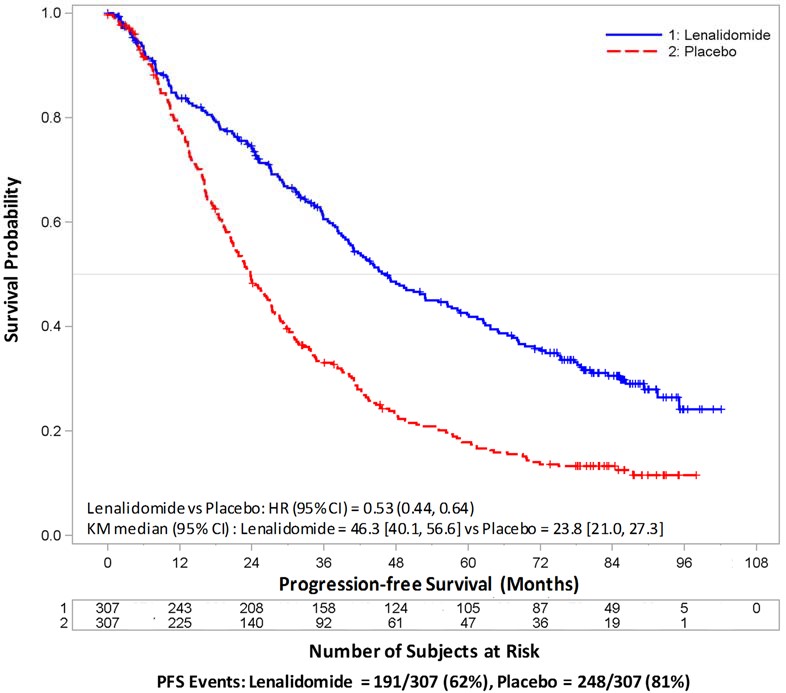
Auto-HSCT = autologous hematopoietic stem cell transplantation; CI = confidence interval; HR = hazard ratio; ITT = intent to treat; KM = Kaplan- Meier; NE = not estimable; PFS = progression-free survival; vs = versus.
Randomized, Open-Label Clinical Studies in Patients with MM After At Least One Prior Therapy
Two randomized studies (Studies 1 and 2) were conducted to evaluate the efficacy and safety of lenalidomide capsules. These multicenter, multinational, double-blind, placebo- controlled studies compared lenalidomide plus oral pulse high-dose dexamethasone therapy to dexamethasone therapy alone in patients with MM who had received at least one prior treatment. These studies enrolled patients with absolute neutrophil counts (ANC) ≥ 1000/mm3, platelet counts ≥ 75,000/mm3, serum creatinine ≤ 2.5 mg/dL, serum SGOT/AST or SGPT/ALT ≤ 3 x upper limit of normal (ULN), and serum direct bilirubin ≤ 2 mg/dL.
In both studies, patients in the lenalidomide/dexamethasone group took 25 mg of lenalidomide capsules orally once daily on Days 1 to 21 and a matching placebo capsule once daily on Days 22 to 28 of each 28-day cycle. Patients in the placebo/dexamethasone group took 1 placebo capsule on Days 1 to 28 of each 28-day cycle. Patients in both treatment groups took 40 mg of dexamethasone orally once daily on Days 1 to 4, 9 to 12, and 17 to 20 of each 28-day cycle for the first 4 cycles of therapy.
The dose of dexamethasone was reduced to 40 mg orally once daily on Days 1 to 4 of each 28-day cycle after the first 4 cycles of therapy. In both studies, treatment was to continue until disease progression.
In both studies, dose adjustments were allowed based on clinical and laboratory findings. Sequential dose reductions to 15 mg daily, 10 mg daily and 5 mg daily were allowed for toxicity [see Dosage and Administration (2.1)].
Table 16 summarizes the baseline patient and disease characteristics in the two studies. In both studies, baseline demographic and disease-related characteristics were comparable between the lenalidomide/dexamethasone and placebo/dexamethasone groups.
Table 16: Baseline Demographic and Disease-Related Characteristics – MM Studies 1 and 2
|
Study 1 |
Study 2 | |||
|
Lenalidomide Capsules**/Dex** |
Placebo/Dex |
Lenalidomide Capsules**/Dex** |
Placebo/Dex | |
|
Patient Characteristics | ||||
|
Age (years) Median |
**** 64 |
**** 62 |
**** 63 |
**** 64 |
|
Sex |
106 (60%) |
104 (59%) |
**** 104 (59%) |
**** 103 (59%) |
|
Race/Ethnicity |
**** 141(80%) |
**** 148 (84%) |
**** 172 (98%) |
**** 175 (100%) |
|
ECOG Performance |
**** 157 (89%) |
**** 168 (95%) |
**** 150 (85%) |
**** 144 (82%) |
|
Disease Characteristics | ||||
|
Multiple Myeloma Stage (Durie-Salmon) |
3% |
3% |
6% |
5% |
|
β2-microglobulin (mg/L)
|
**** 52 (29%) |
**** 51 (29%) |
**** 51 (29%) |
**** 48 (27%) |
|
Number of Prior Therapies | ||||
|
1 |
38% |
38% |
32% |
33% |
|
Types of Prior Therapies | ||||
|
Stem Cell Transplantation |
62% |
61% |
55% |
54% |
|
Thalidomide |
42% |
46% |
30% |
38% |
|
Dexamethasone |
81% |
71% |
66% |
69% |
|
Bortezomib |
11% |
11% |
5% |
4% |
|
Melphalan |
33% |
31% |
56% |
52% |
|
Doxorubicin |
55% |
51% |
56% |
57% |
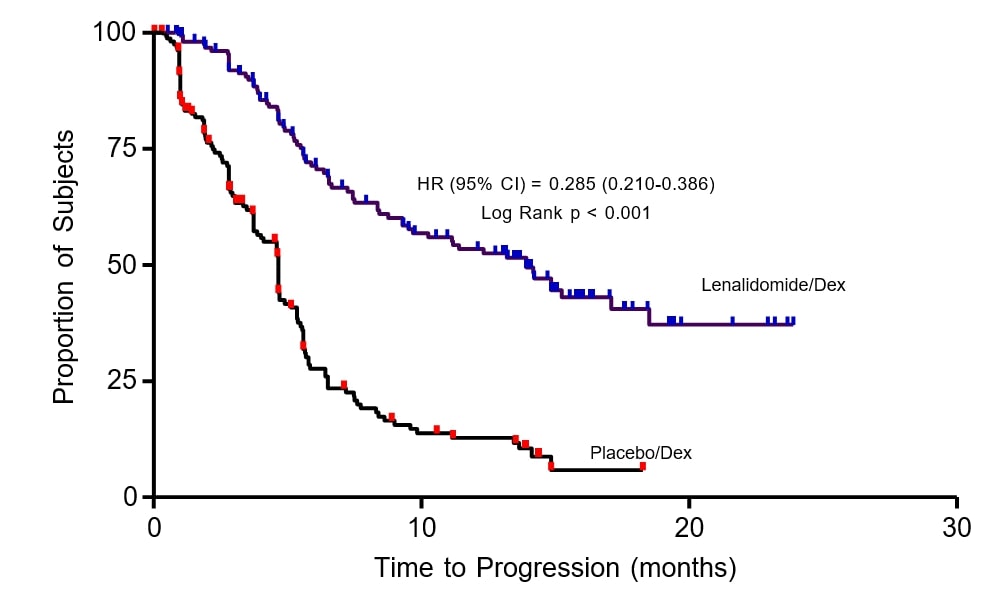
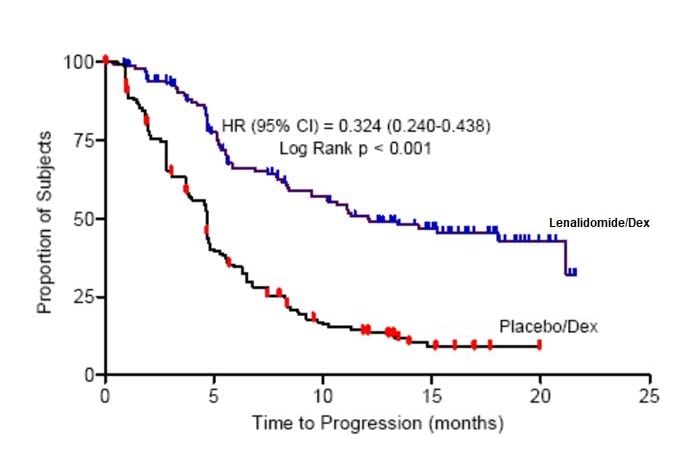
The primary efficacy endpoint in both studies was time to progression (TTP). TTP was defined as the time from randomization to the first occurrence of progressive disease.
Preplanned interim analyses of both studies showed that the combination of lenalidomide/dexamethasone was significantly superior to dexamethasone alone for TTP. The studies were unblinded to allow patients in the placebo/dexamethasone group to receive treatment with the lenalidomide/dexamethasone combination. For both studies, the extended follow- up survival data with crossovers were analyzed. In study 1, the median survival time was 39.4 months (95%CI: 32.9, 47.4) in lenalidomide/dexamethasone group and 31.6 months (95% CI: 24.1, 40.9) in placebo/dexamethasone group, with a hazard ratio of 0.79 (95% CI: 0.61-1.03). In study 2, the median survival time was 37.5 months (95%CI: 29.9, 46.6) in lenalidomide/dexamethasone group and 30.8 months (95%CI: 23.5, 40.3) in placebo/dexamethasone group, with a hazard ratio of 0.86 (95% CI: 0.65-1.14).
Table 17: TTP Results in MM Study 1 and Study 2
|
Study 1 |
Study 2 | |||
|
Lenalidomide Capsules/Dex******** |
Placebo/Dex |
Lenalidomide Capsules /Dex**** |
Placebo/Dex | |
|
TTP | ||||
|
**** Events n (%) |
**** 73 (41) |
**** 120 (68) |
**** 68 (39) |
**** 130 (74) |
|
**** Median TTP in months [95% CI] |
13.9 |
4.7 |
12.1 |
4.7 |
|
Hazard Ratio [95% CI] |
0.285 |
0.324 | ||
|
Log-rank Test p-value 3 |
**** <0.001 |
**** <0.001 | ||
|
Response | ||||
|
Complete Response (CR) n (%) |
**** 23 (13) |
**** 1 (1) |
**** 27 (15) |
**** 7 (4) |
|
Partial Response (RR/PR) n (%) |
**** 84 (48) |
**** 33 (19) |
**** 77 (44) |
**** 34 (19) |
|
Overall Response n (%) |
107 (61) |
34 (19) |
104 (59) |
41 (23) |
|
p-value |
<0.001 |
<0.001 | ||
|
Odds Ratio [95% CI] |
6.38 |
4.72 |
Kaplan-Meier Estimate of Time to Progression — MM Study 1
Kaplan-Meier Estimate of Time to Progression — MM Study 2
14.2 Myelodysplastic Syndromes (MDS) with a Deletion 5q Cytogenetic
Abnormality
The efficacy and safety of lenalidomide were evaluated in patients with transfusion-dependent anemia in low- or intermediate-1- risk MDS with a 5q (q31-33) cytogenetic abnormality in isolation or with additional cytogenetic abnormalities, at a dose of 10 mg once daily or 10 mg once daily for 21 days every 28 days in an open-label, single-arm, multi-center study. The major study was not designed nor powered to prospectively compare the efficacy of the 2 dosing regimens. Sequential dose reductions to 5 mg daily and 5 mg every other day, as well as dose delays, were allowed for toxicity [Dosage and Administration (2.2)].
This major study enrolled 148 patients who had RBC transfusion dependent anemia. RBC transfusion dependence was defined as having received ≥ 2 units of RBCs within 8 weeks prior to study treatment. The study enrolled patients with absolute neutrophil counts (ANC) ≥ 500/mm3, platelet counts ≥ 50,000/mm3, serum creatinine ≤ 2.5 mg/dL, serum SGOT/AST or SGPT/ALT ≤ 3 x upper limit of normal (ULN), and serum direct bilirubin ≤ 2 mg/dL. Granulocyte colony- stimulating factor was permitted for patients who developed neutropenia or fever in association with neutropenia. Baseline patient and disease-related characteristics are summarized in Table 18.
Table 18: Baseline Demographic and Disease-Related Characteristics in the MDS Study
|
** Overall (N=148)** | ||
|
Age (years) | ||
|
Median |
71 | |
|
Min, Max |
37, 95 | |
|
Gender |
n |
(%) |
|
Male |
51 |
(34.5) |
|
Female |
97 |
(65.5) |
|
Race |
n |
(%) |
|
White |
143 |
(96.6) |
|
Other |
5 |
(3.4) |
|
Duration of MDS (years) | ||
|
Median |
2.5 | |
|
Min, Max |
0.1, 20.7 | |
|
Del 5 (q31-33) Cytogenetic Abnormality |
n |
(%) |
|
Yes |
148 |
(100) |
|
Other cytogenetic abnormalities |
37 |
(25.2) |
|
IPSS Score****a |
n |
(%) |
|
Low (0) |
55 |
(37.2) |
|
Intermediate-1 (0.5-1.0) |
65 |
(43.9) |
|
Intermediate-2 (1.5-2.0) |
6 |
(4.1) |
|
High (≥2.5) |
2 |
(1.4) |
|
Missing |
20 |
(13.5) |
|
FAB Classificationb from central review |
n |
(%) |
|
RA |
77 |
(52) |
|
RARS |
16 |
(10.8) |
|
RAEB |
30 |
(20.3) |
|
CMML |
3 |
(2) |
a IPSS Risk Category: Low (combined score = 0), Intermediate-1 (combined score = 0.5 to 1), Intermediate-2 (combined score = 1.5 to 2.0), High (combined score ≥ 2.5); Combined score = (Marrow blast score + Karyotype score + Cytopenia score).
b French-American-British (FAB) classification of MDS.
The frequency of RBC transfusion independence was assessed using criteria modified from the International Working Group (IWG) response criteria for MDS. RBC transfusion independence was defined as the absence of any RBC transfusion during any consecutive “rolling” 56 days (8 weeks) during the treatment period.
Transfusion independence was seen in 99/148 (67%) patients (95% CI [59, 74]). The median duration from the date when RBC transfusion independence was first declared (i.e., the last day of the 56-day RBC transfusion-free period) to the date when an additional transfusion was received after the 56-day transfusion- free period among the 99 responders was 44 weeks (range of 0 to >67 weeks). Ninety percent of patients who achieved a transfusion benefit did so by completion of three months in the study.
RBC transfusion independence rates were unaffected by age or gender.
The dose of lenalidomide was reduced or interrupted at least once due to an adverse event in 118 (79.7%) of the 148 patients; the median time to the first dose reduction or interruption was 21 days (mean, 35.1 days; range, 2 to 253 days), and the median duration of the first dose interruption was 22 days (mean, 28.5 days; range, 2 to 265 days). A second dose reduction or interruption due to adverse events was required in 50 (33.8%) of the 148 patients. The median interval between the first and second dose reduction or interruption was 51 days (mean, 59.7 days; range, 15 to 205 days) and the median duration of the second dose interruption was 21 days (mean, 26 days; range, 2 to 148 days).
14.3 Mantle Cell Lymphoma
A multicenter, single-arm, open-label trial of single-agent lenalidomide was conducted to evaluate the safety and efficacy of lenalidomide in patients with mantle cell lymphoma who have relapsed after or were refractory to bortezomib or a bortezomib-containing regimen. Patients with a creatinine clearance ≥60 mL/min were given lenalidomide at a dose of 25 mg once daily for 21 days every 28 days. Patients with a creatinine clearance ≥30 mL/min and <60 mL/min were given lenalidomide at a dose of 10 mg once daily for 21 days every 28 days. Treatment was continued until disease progression, unacceptable toxicity, or withdrawal of consent.
The trial included patients who were at least 18 years of age with biopsy- proven MCL with measurable disease by CT scan. Patients were required to have received prior treatment with an anthracycline or mitoxantrone, cyclophosphamide, rituximab, and bortezomib, alone or in combination. Patients were required to have documented refractory disease (defined as without any response of PR or better during treatment with bortezomib or a bortezomib- containing regimen), or relapsed disease (defined as progression within one year after treatment with bortezomib or a bortezomib-containing regimen). At enrollment patients were to have an absolute neutrophil counts (ANC) ≥1500/ mm3, platelet counts ≥ 60,000/mm3, serum SGOT/AST or SGPT/ALT ≤3x upper limit of normal (ULN) unless there was documented evidence of liver involvement by lymphoma, serum total bilirubin ≤1.5 x ULN except in cases of Gilbert’s syndrome or documented liver involvement by lymphoma, and calculated creatinine clearance (Cockcroft-Gault formula) ≥30 mL/min.
The median age was 67 years (43-83), 81% were male and 96% were Caucasian. The table below summarizes the baseline disease-related characteristics and prior anti- lymphoma therapy in the Mantle Cell Lymphoma trial.
Table 19: Baseline Disease-related Characteristics and Prior Anti –Lymphoma Therapy in Mantle Cell Lymphoma Trial
|
Baseline Disease Characteristics and Prior Anti - Lymphoma Treatment |
Total Patients (N=134) |
|
ECOG Performance Statusa n (%) |
43 (32) |
|
Advanced MCL Stage, n (%) |
27 (20) |
|
High or Intermediate MIPI Scoreb, n (%) |
90 (67) |
|
High Tumor Burdenc, n (%) |
77 (57) |
|
Bulky Diseased, n (%) |
44 (33) |
|
Extranodal Disease, n (%) |
101 (75) |
|
Number of Prior Systemic Anti-Lymphoma Therapies, n (%) Median (range) |
4 (2, 10) |
|
Number of Subjects Who Received Prior Regimen Containing, n (%):
Anthracycline/mitoxantrone |
133 (99) |
|
Refractory to Prior Bortezomib, n (%) |
81 (60) |
|
Refractory to Last Prior Therapy, n (%) |
74 (55) |
|
Prior Autologous Bone Marrow or Stem Cell Transplant, n (%) |
39 (29) |
a ECOG = Eastern Cooperative Oncology Group.
b MIPI = MCL International Prognostic Index.
c High tumor burden is defined as at least one lesion that is ≥5 cm in diameter or 3 lesions that are ≥3 cm in diameter.
d Bulky disease is defined as at least one lesion that is ≥7cm in the longest diameter.
The efficacy endpoints in the MCL trial were overall response rate (ORR) and duration of response (DOR). Response was determined based on review of radiographic scans by an independent review committee according to a modified version of the International Workshop Lymphoma Response Criteria (Cheson, 1999). The DOR is defined as the time from the initial response (at least PR) to documented disease progression. The efficacy results for the MCL population were based on all evaluable patients who received at least one dose of study drug and are presented in Table 20. The median time to response was 2.2 months (range 1.8 to 13 months).
Table 20: Response Outcomes in the Pivotal Mantle Cell Lymphoma Trial
|
Response Analyses (N = 133) |
N (%) |
95% CI |
|
Overall Response Rate (IWRC) (CR + CRu +PR) |
34 (26) |
(18.4, 33.9) |
|
Complete Response (CR + CRu) |
9 (7) |
(3.1, 12.5) |
|
CR |
1 (1) | |
|
CRu |
8 (6) | |
|
Partial Response (PR) |
25 (19) | |
|
Duration of Response (months) |
Median |
95% CI |
|
Duration of Overall Response (CR + CRu + PR) (N = 34) |
16.6 |
(7.7, 26.7) |
14.4 Follicular and Marginal Zone Lymphoma
The efficacy of lenalidomide capsules with rituximab in patients with relapsed or refractory follicular and marginal zone lymphoma was evaluated in the AUGMENT (NCT01938001) and MAGNIFY (NCT01996865) trials.
AUGMENT is a randomized, double-blind, multicenter trial (n=358) in which patients with relapsed or refractory follicular or marginal zone lymphoma were randomized 1:1 to receive lenalidomide capsules and rituximab or rituximab and placebo. AUGMENT included patients diagnosed with Grade 1, 2, or 3a follicular lymphoma, who received at least 1 prior systemic therapy, were refractory or relapsed, not rituximab-refractory, had at least one measurable nodal or extranodal lesion by CT or MRI scan, and had adequate bone marrow, liver, and renal function. Randomization was stratified by follicular versus marginal zone lymphoma, previous rituximab therapy, and time since other anti-lymphoma therapy. In AUGMENT, lenalidomide capsule was administered orally 20 mg once daily for Days 1 to 21 of repeating 28-day cycles for a maximum of 12 cycles or until unacceptable toxicity. The dose of rituximab was 375 mg/m2 every week in Cycle 1 (Days 1, 8, 15, and 22) and on Day 1 of every 28-day cycle from Cycles 2 through 5. All dosage calculations for rituximab were based on the patient’s body surface area (BSA), using actual patient weight. Dose adjustments for lenalidomide capsules were allowed based on clinical and laboratory findings. A patient with moderate renal insufficiency (≥30 to <60 mL/minute) received a lower lenalidomide capsule starting dose of 10 mg daily on the same schedule. After 2 cycles, the lenalidomide capsules dose could be increased to 15 mg once daily on Days 1 to 21 of each 28-day cycle if the patient tolerated the medication.
MAGNIFY is an open-label, multicenter trial (n=232) in which patients with relapsed or refractory follicular, marginal zone, or mantle cell lymphoma received 12 induction cycles of lenalidomide capsules and rituximab. MAGNIFY included patients diagnosed with Grade 1, 2,3a, 3b follicular (including transformed), marginal zone, or mantle cell lymphoma Stage I to IV who were previously treated for their lymphoma, had been refractory or had a relapse after their last treatment, had at least one measurable nodal or extranodal lesion by CT or MRI scan, and had adequate bone marrow, liver, and renal function. Patients refractory to rituximab were also included. The information from the subjects who received at least 1 dose of initial therapy in the first 12 induction cycles (n=222) in the MAGNIFY trial was included in the evaluation of the efficacy of lenalidomide/rituximab in patients with relapsed or refractory follicular and marginal zone lymphoma. In MAGNIFY, lenalidomide capsule 20 mg was given on Days 1-21 of repeated 28-day cycles for up to 12 cycles or until unacceptable toxicity, progression, or withdrawal of consent. The dose of rituximab was 375 mg/m2 every week in Cycle 1 (Days 1, 8, 15, and 22) and on Day 1 of every other 28-day cycle (Cycles 3,5,7,9, and 11) up to 12 cycles therapy. All dosage calculations for rituximab were based on the patient BSA and actual weight. Dose adjustments were allowed based on clinical and laboratory findings.
The demographic and disease-related baseline characteristics in the AUGMENT and MAGNIFY trials are shown in the following table.
Table 21: Baseline Demographics and Disease-Related Characteristics of Patients with FL and MZL in AUGMENT and MAGNIFY Trials
|
********Parameter |
AUGMENT Trial |
MAGNIFY Trial | |
|
Lenalidomide Capsules + Rituximab |
Rituximab + Placebo |
Lenalidomide Capsules + Rituximab | |
|
Age (years) | |||
|
Median (Max, Min) |
64 (26, 86) |
62 (35, 88) |
65 (35, 91) |
|
Age distribution, n (%) | |||
|
<65 years |
96 (54) |
107 (59) |
103 (46) |
|
≥65 years |
82 (46) |
73 (41) |
119 (54) |
|
Sex, n (%) | |||
|
Male |
75 (42) |
97 (54) |
122 (55) |
|
Female |
103 (58) |
83 (46) |
100 (45) |
|
Race | |||
|
White |
118 (66) |
115 (64) |
206 (93) |
|
Other races |
54 (30) |
64 (36) |
14 (6) |
|
Not collected or reported |
6 (3) |
1 (0.6) |
2 (1) |
|
Body Surface Area (BSA, m2) | |||
|
Median (Max, Min) |
1.8 (1.4, 3.1) |
1.8 (1.3, 2.7) |
2 (1.3, 2.6) |
|
Disease Type FL or MZL | |||
|
Follicular lymphoma |
147 (83) |
148 (82) |
177 (80) |
|
Marginal zone lymphoma |
31 (17) |
32 (18) |
45 (20) |
|
MZL subtype at diagnosis (investigator), n (%) | |||
|
MALT |
14 (45) |
16 (50) |
10 (22) |
|
Nodal |
8 (26) |
10 (31) |
25 (56) |
|
Splenic |
9 (29) |
6 (19) |
10 (22) |
|
FL stage at diagnosis (investigator), n (%) | |||
|
FL Grade 1-2 |
125 (85) |
123 (83) |
149 (84) |
|
FL Grade 3a |
22 (15) |
25 (17) |
28 (16) |
|
FLIPI score at baseline (calculated), n (%) |
Not Collected | ||
|
Low risk (0,1) |
52 (29) |
67 (37) | |
|
Intermediate risk (2) |
55 (31) |
58 (32) | |
|
High risk (≥3) |
69 (39) |
54 (30) | |
|
Missing |
2 (1) |
1 (0.6) | |
|
ECOG score at baseline, n (%) | |||
|
0 |
116 (65) |
128 (71) |
102 (46) |
|
1 |
60 (34) |
50 (28) |
113 (51) |
|
2 |
2 (1) |
2 (1) |
7 (3) |
|
High tumor burdena at baseline, n (%) | |||
|
Yes |
97 (54) |
86 (48) |
148 (67) |
|
No |
81 (46) |
94 (52) |
74 (33) |
|
Number of prior systemic antilymphoma therapies | |||
|
1 |
102 (57) |
97 (54) |
94 (42)b |
|
76 (43) |
83 (46) |
128 (58) |
Data Cutoff: 22 June 2018 (AUGMENT) and 1 May 2017 (MAGNIFY).
a Defined by GELF criteria.
b Patient had either 0 (n=2) or 1 prior systemic therapy.
ECOG = Eastern Cooperative Oncology Group; FLIPI = follicular lymphoma international prognostic index
In AUGMENT, efficacy was established in the intent-to-treat (ITT) population based on progression-free survival by Independent Review Committee using modified 2007 International Working Group response criteria. Efficacy results are summarized in Table 22.
Table 22: Efficacy Results for Patients in the AUGMENT Trial (ITT FL and MZL Population)
|
Parameter |
Lenalidomide Capsules + Rituximab**** |
Rituximab** + Placebo****** |
|
PFS | ||
|
Patients with event, n (%) |
68 (38.2) |
115 (63.9) |
|
Death |
6 (8.8) |
2 (1.7) |
|
Progression of disease |
62 (91.2) |
113 (98.3) |
|
PFS, median a [95% CI] (months) |
39.4 [ 22.9, NE] |
14.1 [11.4, 16.7] |
|
HRb****[95% CI] |
0.46 [ 0.34, 0.62] | |
|
p-value c |
<0.0001 | |
|
**Objective response (CR+PR) , n(%) [95% CI]**d |
138 (77.5) [70.7, 83.4] |
96 (53.3) [45.8, 60.8] |
a Median estimate is from Kaplan-Meier analysis.
b hazard ratio and its CI were estimated from Cox proportional hazard model adjusting for the stratification 3: previous rituximab treatment (yes, no), time since last antilymphoma therapy (≤ 2, > 2 years), and disease histology (FL, MZL).
c p-value from log-rank test stratified by 3 factors noted above: previous rituximab treatment (yes, no), time since last antilymphoma therapy (≤ 2, > 2 years), and disease histology (FL, MZL).
d Exact confidence interval for binomial distribution.
Kaplan-Meier Curves of Progression-free Survival by IRC Assessment Between Arms in AUGMENT Trial (ITT FL and MZL Population)
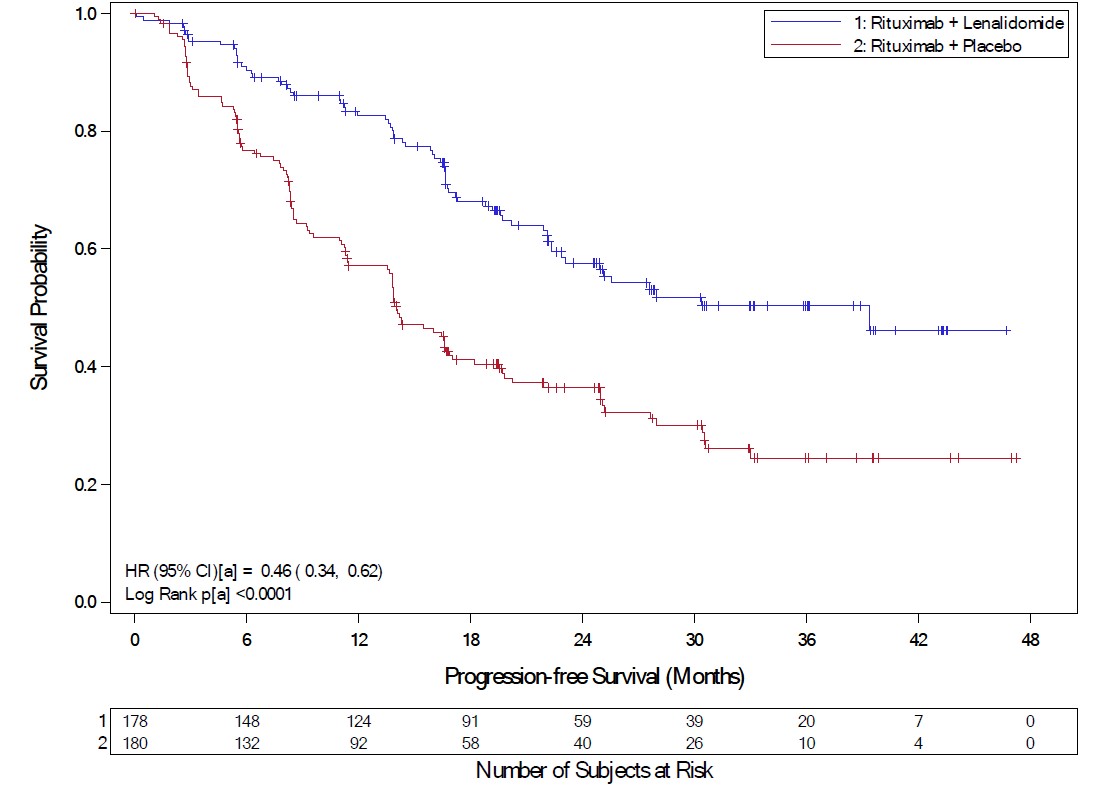
a = Stratification factors included: previous rituximab treatment (y/n), time since last anti-lymphoma therapy (≤2 years, >2years), and disease histology (FL or MZL). CI = confidence interval; HR = hazard ratio; KM = Kaplan-Meier; PFS = progression-free survival
Follicular Lymphoma
In AUGMENT, the objective response by IRC assessment for patients with follicular lymphoma was 80% (118/147) [95% CI: 73%, 86%]) in lenalidomide capsules with rituximab arm compared to 55% (82/148) [95% CI: 47, 64] in control arm.
In MAGNIFY, the overall response by investigator assessment was 59% (104/177) [95% CI: 51, 66] for patients with follicular lymphoma. Median duration of response was not reached with a median follow-up time of 7.9 months [95% CI: 4.6, 9.2].
Marginal Zone Lymphoma
In AUGMENT, the objective response by IRC assessment for patients with marginal zone lymphoma was 65% (20/31) [95% CI: 45%, 81%] in lenalidomide capsules with rituximab arm compared to 44% (14/32) [95% CI: 26%, 62%] in control arm.
In MAGNIFY, the overall response by investigator assessment was 51% (23/45) [95% CI: 36, 66] for patients with marginal zone lymphoma. Median duration of response was not reached with a median follow-up time of 11.5 months [95% CI: 8.0, 18.9].