
Maraviroc
These highlights do not include all the information needed to use MARAVIROC TABLETS safely and effectively. See full prescribing information for MARAVIROC TABLETS. MARAVIROC tablets, for oral use Initial U.S. Approval: 2007
f7b9c67b-10c6-45b5-b2a8-952cd350b0b1
HUMAN PRESCRIPTION DRUG LABEL
Aug 18, 2023
i3 Pharmaceuticals, LLC
DUNS: 080127275
Products 2
Detailed information about drug products covered under this FDA approval, including NDC codes, dosage forms, ingredients, and administration routes.
Maraviroc
Product Details
FDA regulatory identification and product classification information
FDA Identifiers
Product Classification
Product Specifications
INGREDIENTS (11)
Maraviroc
Product Details
FDA regulatory identification and product classification information
FDA Identifiers
Product Classification
Product Specifications
INGREDIENTS (11)
Drug Labeling Information
WARNINGS AND PRECAUTIONS SECTION
5 WARNINGS AND PRECAUTIONS
5.1 Hepatotoxicity
Hepatotoxicity with allergic features including life-threatening events has been reported in clinical trials and postmarketing. Severe rash or evidence of systemic allergic reaction including drug-related rash with fever, eosinophilia, elevated IgE, or other systemic symptoms have been reported in conjunction with hepatotoxicity [see Warnings and Precautions (5.2)] . These events occurred approximately 1 month after starting treatment. Among reported cases of hepatitis, some were observed in the absence of allergic features or with no pre-existing hepatic disease.
Appropriate laboratory testing including ALT, AST, and bilirubin should be conducted prior to initiating therapy with Maraviroc and at other time points during treatment as clinically indicated. Hepatic laboratory parameters should be obtained in any patient who develops rash, or signs or symptoms of hepatitis, or allergic reaction. Discontinuation of Maraviroc should be considered in any patient with signs or symptoms of hepatitis, or with increased liver transaminases combined with rash or other systemic symptoms.
When administering Maraviroc to patients with pre-existing liver dysfunction or who are co-infected with hepatitis B and/or C virus, additional monitoring may be warranted. The safety and efficacy of Maraviroc have not been specifically studied in patients with significant underlying liver disorders.
5.2 Severe Skin and Hypersensitivity Reactions
Severe, potentially life-threatening skin and hypersensitivity reactions have been reported in patients taking Maraviroc, in most cases concomitantly with other drugs associated with these reactions. These include cases of Stevens- Johnson syndrome (SJS), toxic epidermal necrolysis (TEN), and drug rash with eosinophilia and systemic symptoms (DRESS) [see Adverse Reactions (6.2)] . The cases were characterized by features including rash, constitutional findings, and sometimes organ dysfunction, including hepatic failure. Discontinue Maraviroc and other suspected agents immediately if signs or symptoms of severe skin or hypersensitivity reactions develop (including, but not limited to, severe rash or rash accompanied by fever, malaise, muscle or joint aches, blisters, oral lesions, conjunctivitis, facial edema, lip swelling, eosinophilia). Delay in stopping treatment with Maraviroc or other suspect drugs after the onset of rash may result in a life-threatening reaction. Clinical status, including liver aminotransferases, should be monitored and appropriate therapy initiated.
5.3 Cardiovascular Events
Eleven subjects (1.3%) who received Maraviroc had cardiovascular events, including myocardial ischemia and/or infarction, during the Phase 3 trials in treatment‑experienced subjects (total exposure 609 patient‑years [300 on Maraviroc once daily + 309 on Maraviroc twice daily]), while no subjects who received placebo had such events (total exposure 111 patient‑years). These subjects generally had cardiac disease or cardiac risk factors prior to use of Maraviroc, and the relative contribution of Maraviroc to these events is not known.
In the Phase 2b/3 trial in treatment‑naive adult subjects, 3 subjects (0.8%) who received Maraviroc had events related to ischemic heart disease and 5 subjects (1.4%) who received efavirenz had such events (total exposure 506 and 508 patient‑years for Maraviroc and efavirenz, respectively).
When Maraviroc was administered to healthy volunteers at doses higher than the recommended dose, symptomatic postural hypotension was seen at a greater frequency than in placebo. However, when Maraviroc was given at the recommended dose in HIV-1–infected adult subjects in Phase 3 trials, postural hypotension was seen at a rate similar to placebo (approximately 0.5%).
Patients with cardiovascular comorbidities, risk factors for postural hypotension, or receiving concomitant medication known to lower blood pressure, could be at increased risk of cardiovascular adverse events triggered by postural hypotension. Additional monitoring may be warranted.
Postural Hypotension in Patients with Renal Impairment
An increased risk of postural hypotension may occur in patients with severe renal insufficiency or in those with ESRD due to increased maraviroc exposure in some patients. Maraviroc should be used in patients with severe renal impairment or ESRD only if they are not receiving a concomitant potent CYP3A inhibitor or inducer. However, the use of Maraviroc in these patients should only be considered when no alternative treatment options are available. If adult patients with severe renal impairment or ESRD experience any symptoms of postural hypotension while taking 300 mg twice daily, the dose should be reduced to 150 mg twice daily [see Dosage and Administration (2.5)] .
5.4 Immune Reconstitution Syndrome
Immune reconstitution syndrome has been reported in patients treated with combination antiretroviral therapy, including Maraviroc. During the initial phase of combination antiretroviral treatment, patients whose immune systems respond may develop an inflammatory response to indolent or residual opportunistic infections (such as infection with Mycobacterium aviuminfection, cytomegalovirus, Pneumocystis jirovecii pneumonia[PCP], tuberculosis, or reactivation of Herpes simplexand Herpes zoster), which may necessitate further evaluation and treatment.
Autoimmune disorders (such as Graves’ disease, polymyositis, and Guillain- Barré syndrome) have also been reported to occur in the setting of immune reconstitution; however, the time to onset is more variable, and can occur many months after initiation of treatment.
5.5 Potential Risk of Infection
Maraviroc antagonizes the CCR5 co-receptor located on some immune cells, and therefore could potentially increase the risk of developing infections. The overall incidence and severity of infection, as well as AIDS-defining category C infections, were comparable in the treatment groups during the Phase 3 adult treatment‑experienced trials of Maraviroc. While there was a higher rate of certain upper respiratory tract infections reported in the treatment arm receiving Maraviroc compared with placebo (23% versus 13%), there was a lower rate of pneumonia (2% versus 5%) reported in subjects receiving Maraviroc. A higher incidence of Herpes virus infections (11 per 100 patient‑years) was also reported in the treatment arm receiving Maraviroc when adjusted for exposure compared with placebo (8 per 100 patient‑years).
In the Phase 2b/3 trial in treatment‑naive adult subjects, the incidence of AIDS-defining Category C events when adjusted for exposure was 1.8 for Maraviroc compared with 2.4 for efavirenz per 100 patient‑years of exposure.
Patients should be monitored closely for evidence of infections while receiving Maraviroc
5.6 Potential Risk of Malignancy
While no increase in malignancy has been observed with Maraviroc, due to this drug’s mechanism of action, it could affect immune surveillance and lead to an increased risk of malignancy.
The exposure-adjusted rate for malignancies per 100 patient‑years of exposure in adult treatment‑experienced trials was 4.6 for Maraviroc compared with 9.3 on placebo. In treatment‑naive adult subjects, the rates were 1.0 and 2.4 per 100 patient‑years of exposure for Maraviroc and efavirenz, respectively.
Long-term follow-up is needed to more fully assess this risk.
• Hepatotoxicity accompanied by severe rash or systemic allergic reaction, including potentially life- threatening events, has been reported. Hepatic laboratory parameters including alanine aminotransferase (ALT), aspartate aminotransferase (AST), and bilirubinshould be obtained prior to starting Maraviroc and at other time points during treatment as clinically indicated. If rash or symptoms or signs of hepatitis or allergic reaction develop, hepatic laboratory parameters should be monitored and discontinuation of treatment should be considered. When administering Maraviroc to patients with pre-existing liver dysfunction or who are co-infected with hepatitis B and/or C virus, additional monitoring may be warranted. ( 5.1)
• Severe and potentially life-threatening skin and hypersensitivity reactions have been reported in patients taking Maraviroc. This includes cases of Stevens-Johnson syndrome, hypersensitivity reaction, and toxic epidermal necrolysis. Immediately discontinue Maraviroc and other suspected agents if signs or symptoms of severe skin or hypersensitivity reactions develop and monitor clinical status, including liver aminotransferases, closely. ( 5.2)
• More cardiovascular events, including myocardial ischemia and/or infarction, were observed in treatment-experienced subjects who received Maraviroc. Additional monitoring may be warranted. ( 5.3)
• If patients with severe renal impairment or ESRD receiving Maraviroc (without concomitant CYP3A inducers or inhibitors) experience postural hypotension, the dose of Maraviroc should be reduced from 300 mg twice daily to 150 mg twice daily. ( 5.3)
ADVERSE REACTIONS SECTION
6 ADVERSE REACTIONS
The following adverse reactions are discussed in other sections of the labeling:
- Hepatotoxicity [see Boxed Warning, Warnings and Precautions (5.1)]
- Severe Skin and Hypersensitivity Reactions [see Warnings and Precautions (5.2)]
- Cardiovascular Events [see Warnings and Precautions (5.3)]
6.1 Clinical Trials Experience
Because clinical trials are conducted under widely varying conditions, adverse reaction rates observed in the clinical trials of a drug cannot be directly compared with rates in the clinical trials of another drug and may not reflect the rates observed in practice.
Clinical Trials Experience in Adult Subjects
Treatment‑Experienced Subjects:The safety profile of Maraviroc is primarily based on 840 HIV-1infected subjects who received at least 1 dose of Maraviroc during two Phase 3 trials. A total of 426 of these subjects received the indicated twice‑daily dosing regimen.
Assessment of treatment‑emergent adverse events is based on the pooled data from 2 trials in subjects with CCR5-tropic HIV‑1 (A4001027 and A4001028). The median duration of therapy with Maraviroc for subjects in these trials was 48 weeks, with the total exposure on Maraviroc twice daily at 309 patient‑years versus 111 patient‑years on placebo each administered with optimized background therapy (OBT). The population was 89% male and 84% white, with mean age of 46 years (range: 17 to 75 years). Subjects received dose equivalents of 300 mg maraviroc once or twice daily.
The most common adverse events reported with twice‑daily therapy with Maraviroc with frequency rates higher than placebo, regardless of causality, were upper respiratory tract infections, cough, pyrexia, rash, and dizziness. In these 2 trials, the rate of discontinuation due to adverse events was 5% for subjects who received Maraviroc twice daily + OBT as well as those who received placebo + OBT. Most of the adverse events reported were judged to be mild to moderate in severity. The data described below occurred with twice‑daily dosing of Maraviroc.
The total numbers of subjects reporting infections were 233 (55%) and 84 (40%) in the group receiving Maraviroc twice daily and the placebo group, respectively. Correcting for the longer duration of exposure on Maraviroc compared with placebo, the exposure‑adjusted frequency (rate per 100 subject‑years) of these events was 133 for both Maraviroc twice daily and placebo.
Dizziness or postural dizziness occurred in 8% of subjects on either Maraviroc or placebo, with 2 subjects (0.5%) on Maraviroc permanently discontinuing therapy (1 due to syncope, 1 due to orthostatic hypotension) versus 1 subject on placebo (0.5%) permanently discontinuing therapy due to dizziness.
Treatment-emergent adverse events, regardless of causality, from Trials A4001027 and A4001028 are summarized in Table 5. Selected events occurring at greater than or equal to 2% of subjects and at a numerically higher rate in subjects treated with Maraviroc are included; events that occurred at the same or higher rate on placebo are not displayed.
Table 5. Selected Treatment-Emergent Adverse Events (All Causality) ≥2% on Maraviroc (and at a Higher Rate Compared with Placebo) in Trials A4001027 and A4001028 (Pooled Analysis, 48 Weeks)
| ||||
|
** Body System/** Adverse Event |
Maraviroc |
Placebo | ||
|
** (n = 426)** |
** Exposure- Adjusted Rate (per 100 pt- yrs)** |
** (n = 209)** |
** Exposure- Adjusted Rate (per 100 pt- yrs)** | |
|
Eye Disorders | ||||
|
Conjunctivitis |
2 |
3 |
1 |
3 |
|
Ocular infections, inflammations, and associated manifestations |
2 |
3 |
1 |
2 |
|
Gastrointestinal Disorders | ||||
|
Constipation |
6 |
9 |
3 |
6 |
|
General Disorders and Administration Site Conditions | ||||
|
Pyrexia |
13 |
20 |
9 |
17 |
|
Pain and discomfort |
4 |
5 |
3 |
5 |
|
Infections and Infestations | ||||
|
Upper respiratory tract infection |
23 |
37 |
13 |
27 |
|
Herpes infection |
8 |
11 |
4 |
8 |
|
Sinusitis |
7 |
10 |
3 |
6 |
|
Bronchitis |
7 |
9 |
5 |
9 |
|
Folliculitis |
4 |
5 |
2 |
4 |
|
Anogenital warts |
2 |
3 |
1 |
3 |
|
Influenza |
2 |
3 |
0.5 |
1 |
|
Otitis media |
2 |
3 |
0.5 |
1 |
|
Metabolism and Nutrition Disorders | ||||
|
Appetite disorders |
8 |
11 |
7 |
13 |
|
Musculoskeletal and Connective Tissue Disorders | ||||
|
Joint-related signs and symptoms |
7 |
10 |
3 |
5 |
|
Muscle pains |
3 |
4 |
0.5 |
1 |
|
Neoplasms Benign, Malignant, and Unspecified | ||||
|
Skin neoplasms benign |
3 |
4 |
1 |
3 |
|
Nervous System Disorders | ||||
|
Dizziness/postural dizziness |
9 |
13 |
8 |
17 |
|
Paresthesias and dysesthesias |
5 |
7 |
3 |
6 |
|
Sensory abnormalities |
4 |
6 |
1 |
3 |
|
Disturbances in consciousness |
4 |
5 |
3 |
6 |
|
Peripheral neuropathies |
4 |
5 |
3 |
6 |
|
Psychiatric Disorders | ||||
|
Disturbances in initiating and maintaining sleep |
8 |
11 |
5 |
10 |
|
Depressive disorders |
4 |
6 |
3 |
5 |
|
Anxiety symptoms |
4 |
5 |
3 |
7 |
|
Renal and Urinary Disorders | ||||
|
Bladder and urethral symptoms |
5 |
7 |
1 |
3 |
|
Urinary tract signs and symptoms |
3 |
4 |
1 |
3 |
|
Respiratory, Thoracic, and Mediastinal Disorders | ||||
|
Coughing and associated symptoms |
14 |
21 |
5 |
10 |
|
Upper respiratory tract signs and symptoms |
6 |
9 |
3 |
6 |
|
Nasal congestion and inflammations |
4 |
6 |
3 |
5 |
|
Breathing abnormalities |
4 |
5 |
2 |
5 |
|
Paranasal sinus disorders |
3 |
4 |
0.5 |
1 |
|
Skin and Subcutaneous Tissue Disorders | ||||
|
Rash |
11 |
16 |
5 |
11 |
|
Apocrine and eccrine gland disorders |
5 |
7 |
4 |
7.5 |
|
Pruritus |
4 |
5 |
2 |
4 |
|
Lipodystrophies |
3 |
5 |
0.5 |
1 |
|
Erythema |
2 |
3 |
1 |
2 |
|
Vascular Disorders | ||||
|
Vascular hypertensive disorders |
3 |
4 |
2 |
4 |
Laboratory Abnormalities:Table 6 shows the treatment-emergent Grade 3-4 laboratory abnormalities that occurred in greater than 2% of subjects receiving Maraviroc.
Table 6. Maximum Shift in Laboratory Test Values (without Regard to Baseline) ≥2% of Grade 3-4 Abnormalities (ACTG Criteria) in Trials A4001027 and A4001028 (Pooled Analysis, 48 Weeks)
| |||
|
** Laboratory Parameter Preferred Term** |
** Limit** |
** Maraviroc Twice Daily + OBT** % |
Placebo + OBT |
|
Aspartate aminotransferase |
|
4.8 |
2.9 |
|
Alanine aminotransferase |
|
2.6 |
3.4 |
|
Total bilirubin |
|
5.5 |
5.3 |
|
Amylase |
|
5.7 |
5.8 |
|
Lipase |
|
4.9 |
6.3 |
|
Absolute neutrophil count |
<750/mm 3 |
4.3 |
2.4 |
|
ULN = Upper limit of normal; OBT = optimized background therapy. |
Treatment‑Naive Subjects: Treatment-Emergent Adverse Events:Treatment-emergent adverse events, regardless of causality, from Trial A4001026, a double-blind, comparative, controlled trial in which 721 treatment-naive subjects received Maraviroc 300 mg twice daily (n = 360) or efavirenz 600 mg once daily (n = 361) in combination with lamivudine/zidovudine (COMBIVIR) for 96 weeks, are summarized in Table 7. Selected events occurring in greater than or equal to 2% of subjects and at a numerically higher rate in subjects treated with Maraviroc are included; events that occurred at the same or higher rate on efavirenz are not displayed.
Table 7. Selected Treatment-Emergent Adverse Events (All Causality) ≥2% on Maraviroc (and at a Higher Rate Compared with Efavirenz) in Trial A4001026 (96 Weeks)|
** Body System/** ** Adverse Event** |
Maraviroc 300 mg Once Daily + |
** Efavirenz** 600 mg Once Daily + |
|
** Blood and Lymphatic System Disorders** | ||
|
Anemias NEC |
8 |
5 |
|
Neutropenias |
4 |
3 |
|
** Ear and Labyrinth Disorders** | ||
|
Ear disorders NEC |
3 |
2 |
|
** Gastrointestinal Disorders** | ||
|
Flatulence, bloating, and distention |
10 |
7 |
|
Gastrointestinal atonic and hypomotility disorders NEC |
9 |
5 |
|
Gastrointestinal signs and symptoms NEC |
3 |
2 |
|
** General Disorders and Administration Site Conditions** | ||
|
Body temperature perception |
3 |
1 |
|
** Infections and Infestations** | ||
|
Upper respiratory tract infection |
32 |
30 |
|
Bronchitis |
13 |
9 |
|
Herpes infection |
7 |
6 |
|
Bacterial infections NEC |
6 |
3 |
|
Herpes zoster/varicella |
5 |
4 |
|
Tinea infections |
4 |
3 |
|
Lower respiratory tract and lung |
3 |
2 |
|
Neisseria infections |
3 |
0 |
|
Viral infections NEC |
3 |
2 |
|
** Musculoskeletal and Connective Tissue Disorders** | ||
|
Joint-related signs and symptoms |
6 |
5 |
|
** Nervous System Disorders** | ||
|
Paresthesias and dysesthesias |
4 |
3 |
|
Memory loss (excluding dementia) |
3 |
1 |
|
** Renal and Urinary Disorders** | ||
|
Bladder and urethral symptoms |
4 |
3 |
|
** Reproductive System and Breast Disorders** | ||
|
Erection and ejaculation conditions and disorders |
3 |
2 |
|
** Respiratory, Thoracic, and Mediastinal Disorders** | ||
|
Upper respiratory tract signs and symptoms |
9 |
5 |
|
** Skin and Subcutaneous Disorders** | ||
|
Nail and nail bed conditions (excluding infections and |
6 |
2 |
|
Lipodystrophies |
4 |
3 |
|
Acnes |
3 |
2 |
|
Alopecias |
2 |
1 |
Laboratory Abnormalities:
Table 8. Maximum Shift in Laboratory Test Values (without Regard to Baseline) ≥2% of Grade 3-4 Abnormalities (ACTG Criteria) in Trial A4001026 (96 Weeks)
| |||
|
** Laboratory Parameter** Preferred Term |
** Limit** |
** Maraviroc** |
** Efavirenz** |
|
Aspartate aminotransferase |
|
4.0 |
4.0 |
|
Alanine aminotransferase |
|
3.9 |
4.0 |
|
Creatine kinase |
|
3.9 |
4.8 |
|
Amylase |
|
4.3 |
6.0 |
|
Absolute neutrophil count |
<750/mm3 |
5.7 |
4.9 |
|
Hemoglobin |
<7.0 g/dL |
2.9 |
2.3 |
|
ULN = Upper limit of normal. |
Less Common Adverse Events in Clinical Trials:The following adverse events occurred in less than 2% of subjects treated with Maraviroc or at a rate similar to the comparator. These events have been included because of their seriousness and either increased frequency on Maraviroc or are potential risks due to the mechanism of action. Events attributed to the subjects underlying HIV-1 infection are not listed.
Blood and Lymphatic System:Marrow depression and hypoplastic anemia.
Cardiac Disorders:Unstable angina, acute cardiac failure, coronary artery disease, coronary artery occlusion, myocardial infarction, myocardial ischemia.
Hepatobiliary Disorders:Hepatic cirrhosis, hepatic failure, cholestatic jaundice, portal vein thrombosis, jaundice.
Infections and Infestations:Endocarditis, infective myositis, viral meningitis, pneumonia, treponema infections, septic shock, Clostridium difficile colitis, meningitis.
Musculoskeletal and Connective Tissue Disorders:Myositis, osteonecrosis, rhabdomyolysis, blood creatine kinase increased.
Neoplasms Benign, Malignant, and Unspecified (Including Cysts and Polyps):Abdominal neoplasm, anal cancer, basal cell carcinoma, Bowens disease, cholangiocarcinoma, diffuse large B-cell lymphoma, lymphoma, metastases to liver, esophageal carcinoma, nasopharyngeal carcinoma, squamous cell carcinoma, squamous cell carcinoma of skin, tongue neoplasm (malignant stage unspecified), anaplastic large cell lymphomas T- and null-cell types, bile duct neoplasms malignant, endocrine neoplasms malignant and unspecified.
Nervous System Disorders:Cerebrovascular accident, convulsions and epilepsy, tremor (excluding congenital), facial palsy, hemianopia, loss of consciousness, visual field defect.
Clinical Trials Experience in Pediatric Subjects
HIV-1Infected Pediatric Subjects:Trial A4001031 is an open-label trial in which 103 treatment-experienced, CCR5-tropic, HIV-1infected pediatric subjects aged 2 to less than 18 years weighing at least 10 kg received Maraviroc twice daily in combination with OBT. The dose of Maraviroc was based on body surface area (BSA) and on whether the subject was receiving potent CYP3A inhibitors and/or inducers. The median duration of therapy with Maraviroc was 131 weeks with 72% of subjects receiving study treatment for greater than 48 weeks and 62% of subjects receiving study treatment for 96 weeks.
In these 103 children and adolescents, the safety profile through 96 weeks was similar to that for adults. Most of the adverse reactions reported were mild to moderate; severe (Grade 3 and 4) adverse reactions occurred in 2% of subjects. The most common adverse reactions (all grades) reported with twice- daily therapy with Maraviroc were vomiting (12%), abdominal pain (4%), diarrhea (4%), nausea (4%), and dizziness (3%). Three subjects (3%) discontinued due to adverse events.
Maraviroc-related gastrointestinal adverse events through 48 weeks (nausea, vomiting, diarrhea, constipation, and abdominal pain/cramps) were observed more commonly in subjects who received the Maraviroc oral solution (21%) compared with those who received Maraviroc tablets (16%). Subjects were permitted to change formulations after Week 48.
6.2 Postmarketing Experience
The following adverse events have been identified during post-approval use of Maraviroc. Because these reactions are reported voluntarily from a population of uncertain size, it is not always possible to reliably estimate their frequency or establish a causal relationship to drug exposure.
Skin and Subcutaneous Tissue Disorders
Stevens‑Johnson syndrome (SJS), drug rash with eosinophilia and systemic symptoms (DRESS), toxic epidermal necrolysis (TEN).
• The most common adverse events in treatment-experienced adult subjects (greater than 8% incidence) which occurred at a higher frequency compared with placebo are upper respiratory tract infections, cough, pyrexia, rash, and dizziness. ( 6.1)
• The most common adverse events in treatment-naive adult subjects (greater than 8% incidence) which occurred at a higher frequency than the comparator arm are upper respiratory tract infections, bronchitis, flatulence, bloating and distention, upper respiratory tract signs and symptoms, and gastrointestinal atonic and hypomotility disorders. ( 6.1)
• The most common adverse reactions in treatment-experienced pediatric subjects (greater than or equal to 3% incidence) are vomiting, abdominal pain, diarrhea, nausea, and dizziness. ( 6.1)
To report SUSPECTED ADVERSE REACTIONS, contact i3 Pharmaceuticals, LLC at 1-844-874-7353 or FDA at 1-800-FDA-1088 or www.fda.gov/medwatch
DRUG INTERACTIONS SECTION
7 DRUG INTERACTIONS
7.1 Effect of Concomitant Drugs on the Pharmacokinetics of Maraviroc
Maraviroc is metabolized by CYP3A and is also a substrate for P-glycoprotein (P-gp), organic anion-transporting polypeptide (OATP)1B1, and multidrug resistance-associated protein (MRP)2. The pharmacokinetics of maraviroc are likely to be modulated by inhibitors and inducers of CYP3A and P-gp and may be modulated by inhibitors of OATP1B1 and MRP2. Therefore, a dosage adjustment may be required when maraviroc is coadministered with those drugs [see Dosage and Administration ( 2.3, 2.4)] .
Concomitant use of maraviroc and St. John's wort ( Hypericum perforatum) or products containing St. John's wort is not recommended. Coadministration of maraviroc with St. John's wort is expected to substantially decrease maraviroc concentrations and may result in suboptimal levels of maraviroc and lead to loss of virologic response and possible resistance to maraviroc.
Additional drug interaction information is available [see Clinical Pharmacology (12.3)] .
• Coadministration with CYP3A inhibitors, including protease inhibitors (except tipranavir/ritonavir), will increase the concentration of Maraviroc. ( 7.1)
• Coadministration with CYP3A inducers, including efavirenz, may decrease the concentration of Maraviroc. ( 7.1)
• Coadministration with St. John’s wort is not recommended. ( 7.1)
DESCRIPTION SECTION
11 DESCRIPTION
Maraviroc is a selective, slowly reversible, small molecule antagonist of the interaction between human CCR5 and HIV-1 gp120. Blocking this interaction prevents CCR5-tropic HIV-1 entry into cells.
Maraviroc film-coated tablets for oral administration contain 150, or 300 mg of maraviroc and the following inactive ingredients: dibasic calcium phosphate (anhydrous), magnesium stearate, microcrystalline cellulose, and sodium starch glycolate. The film coat (Opadry II Blue [85G20583]) contains FD&C blue # 2 aluminum lake, soya lecithin, polyethylene glycol (macrogol 3350), polyvinyl alcohol, talc, and titanium dioxide.
Maraviroc is chemically described as 4,4-difluoro-N-{(1S)-3-[exo-3-(3-isopropyl-5- methyl-4H-1,2,4-triazol-4-yl)-8-azabicyclo[3.2.1]oct-8-yl]-1- phenylpropyl}cyclohexanecarboxamide.
The molecular formula is C29H41F2N5O and the structural formula is:
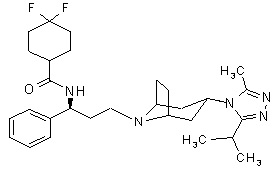
Maraviroc is a white to off-white powder with a molecular weight of 513.68. It is very soluble in methanol and is highly soluble across the physiological pH range (pH 1.0 to 7.5).
HOW SUPPLIED SECTION
16 HOW SUPPLIED/STORAGE AND HANDLING
Maraviroc film-coated tablets are available as follows:
150-mg, and 300-mg tablets are blue, oval, film-coated tablets, debossed “I3” on one side and “24” and “25”, respectively, on the other side.
150-mg tablets: Bottle of 60 tablets (NDC 72319-024-02).
300-mg tablets: Bottle of 60 tablets (NDC 72319-025-02).
Maraviroc film‑coated tablets should be stored at 20 oC to 25 oC (68 oF to 77 oF); excursions permitted between 15 oC and 30 oC (59 oF and 86 oF) [see USP Controlled Room Temperature].
DOSAGE & ADMINISTRATION SECTION
2 DOSAGE AND ADMINISTRATION
2.1 Testing prior to Initiation of Maraviroc
Prior to initiation of Maraviroc for treatment of HIV-1 infection, test all patients for CCR5 tropism using a highly sensitive tropism assay. Maraviroc is recommended for patients with only CCR5-tropic HIV-1 infection. Outgrowth of pre-existing low-level CXCR4- or dual/mixed-tropic HIV-1 not detected by tropism testing at screening has been associated with virologic failure on Maraviroc [see Microbiology (12.4), Clinical Studies (14.1)].
Monitor patients for alanine aminotransferase (ALT), aspartate aminotransferase (AST), and bilirubin prior to initiation of Maraviroc and at other time points during treatment as clinically indicated [see Warnings and Precautions (5.1)].
2.2 General Dosing Recommendations
- Maraviroc tablets are taken twice daily by mouth and may be taken with or without food.
- Maraviroc must be given in combination with other antiretroviral medications.
- The recommended dosage of Maraviroc differs based on concomitant medications due to drug interactions.
2.3 Recommended Dosage in Adult Patients with Normal Renal Function
Table 1 displays oral dosage of Maraviroc based on different concomitant medications [see Drug Interactions (7.1)].
Table 1. Recommended Dosage in Adults
| |
|
** Concomitant Medications** |
Dosage of Maraviroc |
|
Potent cytochrome P450 (CYP)3A inhibitors (with or without a potent CYP3A inducer) * |
150 mg twice daily |
|
Noninteracting concomitant medications † |
300 mg twice daily |
|
Potent and moderate CYP3A inducers (without a potent CYP3A inhibitor) ‡ |
600 mg twice daily |
2.4 Recommended Dosage in Pediatric Patients with Normal Renal Function
The recommended dosage of Maraviroc should be based on body weight (kg) and should not exceed the recommended adult dose. The recommended dosage also differs based on concomitant medications due to drug interactions (Table 2 and Table 3) [see Drug Interactions (7.1), Use in Specific Populations (8.4)].
Before prescribing Maraviroc tablets, assess children for the ability to swallow tablets. If a child is unable to reliably swallow Maraviroc tablets, the oral solution formulation should be prescribed.
The recommended oral dosage of Maraviroc tablets in pediatric patients aged 2 years and older weighing at least 10 kg is presented in Table 2.
Table 2. Recommended Dosage in Pediatric Patients Aged 2 Years and Older Weighing at Least 10 kg (Tablets)
| |||||
|
Concomitant Medications |
Dosage of Maraviroc Based on Weight | ||||
|
** 10 kg to** |
** 14 kg to** |
** 20 kg to** |
** 30 kg to** |
≥40 kg | |
|
Potent CYP3A inhibitors (with or without a CYP3A inducer) * |
50 mg twice daily |
50 mg twice daily |
75 mg twice daily |
100 mg twice daily |
150 mg twice daily |
|
Noninteracting concomitant medications † |
150 mg twice daily |
200 mg twice daily |
200 mg twice daily |
300 mg twice daily |
300 mg twice daily |
|
Potent and moderate CYP3A inducers (without a potent CYP3A inhibitor) ‡ |
Not recommended § |
The recommended oral dosage of maraviroc oral solution in pediatric patients weighing at least 10 kg is presented in Table 3.
Table 3. Recommended Dosage in Pediatric Patients Weighing at Least 10 kg (Oral Solution)
| |||||
|
Concomitant Medications |
Dosage (Volume of Solution) of Maraviroc Based on Weight | ||||
|
10 kg to <14 kg |
14 kg to <20 kg |
20 kg to <30 kg |
30 kg to <40 kg |
≥40 kg | |
|
Potent CYP3A inhibitors (with or without a CYP3A inducer) * |
50 mg (2.5 mL) twice daily |
50 mg (2.5 mL) twice daily |
80 mg (4 mL) twice daily |
100 mg (5 mL) twice daily |
150 mg (7.5 mL) twice daily |
|
Noninteracting concomitant medications † |
150 mg (7.5 mL) twice daily |
200 mg (10 mL) twice daily |
200 mg (10 mL) twice daily |
300 mg (15 mL) twice daily |
300 mg (15 mL) twice daily |
|
Potent and moderate CYP3A inducers (without a potent CYP3A inhibitor) ‡ |
Not recommended § |
Administer the oral solution using the appropriate oral dosing syringe: for doses greater than 2.5 mL, use the 10-mL syringe.
2.5 Recommended Dosage in Patients with Renal Impairment
Adult Patients
Table 4 provides dosing recommendations for patients based on renal function and concomitant medications.
Table 4. Recommended Dosage in Adults Based on Renal Function
| |||||
|
Concomitant Medications |
Dosage of Maraviroc Based on Renal Function | ||||
|
** Normal (CrCl >80** |
** Mild (CrCl >50** |
** Moderate (CrCl ≥30 and ≤50 mL/min)** |
** Severe** |
End-Stage Renal Disease on Regular Hemodialysis | |
|
Potent CYP3A inhibitors (with or without a CYP3A inducer) * |
150 mg twice daily |
150 mg twice daily |
150 mg twice daily |
Contraindicated |
Contraindicated |
|
Noninteracting concomitant medications † |
300 mg twice daily |
300 mg twice daily |
300 mg twice daily |
300 mg twice daily |
300 mg twice daily ‡ |
|
Potent and moderate CYP3A inducers (without a potent CYP3A inhibitor) § |
600 mg twice daily |
600 mg twice daily |
600 mg twice daily |
Contraindicated |
Contraindicated |
|
CrCl = Creatinine Clearance. |
Pediatric Patients
There are no data to recommend specific doses of Maraviroc in pediatric patients with mild or moderate renal impairment [see Use in Specific Populations (8.6)] . Additionally, Maraviroc is contraindicated for pediatric patients with severe renal impairment or end- stage renal disease (ESRD) on regular hemodialysis who are receiving potent CYP3A inhibitors or inducers [see Contraindications (4)].
• Prior to initiation of Maraviroc for treatment of HIV-1 infection, test all patients for CCR5 tropism using a highly sensitive tropism assay. ( 2.1)
• Maraviroc tablets are taken twice daily by mouth and may be taken with or without food. Maraviroc must be given in combination with other antiretroviral medications. ( 2.2)
Recommended Dosage in Adult Patients: ( 2.3)
|
Concomitant Medications |
Dosage of Maraviroc |
|
When given with potent cytochrome P450 (CYP)3A inhibitors (with or without potent CYP3A inducers) including PIs (except tipranavir/ritonavir) ( 2.3, 7.1) |
150 mg twice daily |
|
With NRTIs, tipranavir/ritonavir, nevirapine, raltegravir, and other drugs that are not potent CYP3A inhibitors or CYP3A inducers ( 2.3, 7.1) |
300 mg twice daily |
|
With potent and moderate CYP3A inducers including efavirenz (without a potent CYP3A inhibitor) ( 2.3, 7.1) |
600 mg twice daily |
A more complete list of coadministered drugs is listed in Dosage and Administration. ( 2)
Recommended Dosage in Pediatric Patients 2 years and older and weighing at Least 10 kg: Administer twice daily. Dosage should be based on body weight (kg) and concomitant medications and should not exceed the recommended adult dose. ( 2.4)
Recommended Dosage in Patients with Renal Impairment: Dose adjustment may be necessary in adult patients with renal impairment. ( 2.5)