
Regulatory Information
HSA regulatory responsibility and product classification details
Regulatory Responsibility
Product Classification
Formulation Information
INJECTION, SOLUTION (RADIOPHARMACEUTICAL)
**4 Dosage regimen and administration** **Important safety instructions** Lutathera is a radiopharmaceutical and should be handled with appropriate safety measures to minimize radiation exposure in accordance with national regulations and/or institutional guidelines (see section Warnings and precautions – _please refer to the Product Insert/Patient Information Leaflet published on HSA for the full drug information_). Waterproof gloves and effective radiation shielding should be used when handling Lutathera (see section Pharmaceutical information – _please refer to the Product Insert/Patient Information Leaflet published on HSA for the full drug information_). Radiopharmaceuticals, including Lutathera, should be used by or under the control of physicians who are qualified by specific training and experience in the safe use and handling of radiopharmaceuticals, and whose experience and training have been approved by the appropriate governmental agency authorized to license the use of radiopharmaceuticals. Pregnancy status of females of reproductive potential must be verified prior to initiating treatment with Lutathera (see section Pregnancy, lactation, females and males of reproductive potential – _please refer to the Product Insert/Patient Information Leaflet published on HSA for the full drug information_). **Patient identification** Before initiating treatment with Lutathera, presence of somatostatin receptor-positive tumors must be confirmed, preferably by somatostatin receptor imaging. **Dosage regimen** **General target population** **Adults** The recommended treatment regimen of Lutathera in adults consists of 4 infusions of 7.4 GBq each. The recommended interval between each infusion is 8 weeks (±1 week). **Premedications and concomitant medications** **Antiemetics** Antiemetics should be administered with sufficient lead time prior to the start of the amino acid solution. Refer to full prescribing information of antiemetics for administration instructions. In case of severe nausea or vomiting during the infusion of the amino acid solution despite administration of a pre-procedure antiemetic, an antiemetic of a different pharmacological class can be administered. **Amino acid solution** For renal protection, an intravenous amino acid solution containing L-lysine and L-arginine must be administered 30 minutes before the start of the Lutathera infusion (see Tables 4-1 and 4-2). The amino acid solution infusion should continue during, and for at least 3 hours after the completion of the Lutathera infusion. Infusion of the amino acid solution and Lutathera through a separate venous access in each of the patient’s arms is the preferred method. However, if two intravenous lines are not possible due to poor venous access or institutional/clinical preference, the amino acid solution and Lutathera may be infused through the same line via a three-way valve, taking into consideration flow rate and maintenance of venous access. The dose of the amino acid solution should not be decreased even if a reduced dose of Lutathera is administered. An amino acid solution containing just L-lysine and L-arginine in the amounts specified in Table 4-1 (e.g., LysaKare®) is considered the medicinal product of choice, due to the lower total volume to be infused and lower osmolality. The amino acid solution can be prepared as a compounded product, in compliance with the hospital’s good preparation practices for sterile medicinal products and according to the composition specified in Table 4-1.  Commercially available amino acid solutions (e.g., LysaKare®) can be used if compliant with the specification listed in Table 4-2. 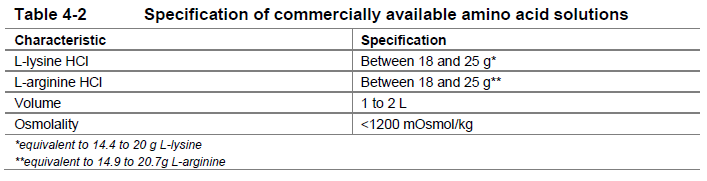 **Treatment monitoring** Before each administration and during treatment with Lutathera, hematology (platelet count, white blood cell count with differential counts and hemoglobin \[Hb\]), kidney function test (serum creatinine and creatinine clearance by Cockcroft-Gault formula) and liver function test (alanine aminotransferase \[ALT\], aspartate aminotransferase \[AST\], serum albumin, INR and bilirubin) should be performed to assess the patient’s condition and adapt the therapeutic protocol if necessary (dose, infusion interval, number of infusions) (see Table 4-3). These laboratory tests should be performed shortly before each administration and between 4 to 6 weeks after each dose of Lutathera. It is also recommended to perform these tests every 4 weeks for at least 3 months after the last infusion of Lutathera and every 6 months thereafter, in order to be able to detect possible delayed adverse drug reactions (ADRs) (see section Adverse drug reactions – _please refer to the Product Insert/Patient Information Leaflet published on HSA for the full drug information_). Dosing may need to be modified based on the tests results as described in Table 4-3 Recommended dose modifications for adverse drug reactions. **Dose modifications for adverse drug reactions** Management of severe or intolerable adverse drug reactions may require temporary dose interruption (extension of the dosing interval from 8 weeks up to 16 weeks), dose reduction, or permanent discontinuation of treatment with Lutathera. Recommended dose modifications of Lutathera for adverse drug reactions are provided in Table 4-3. 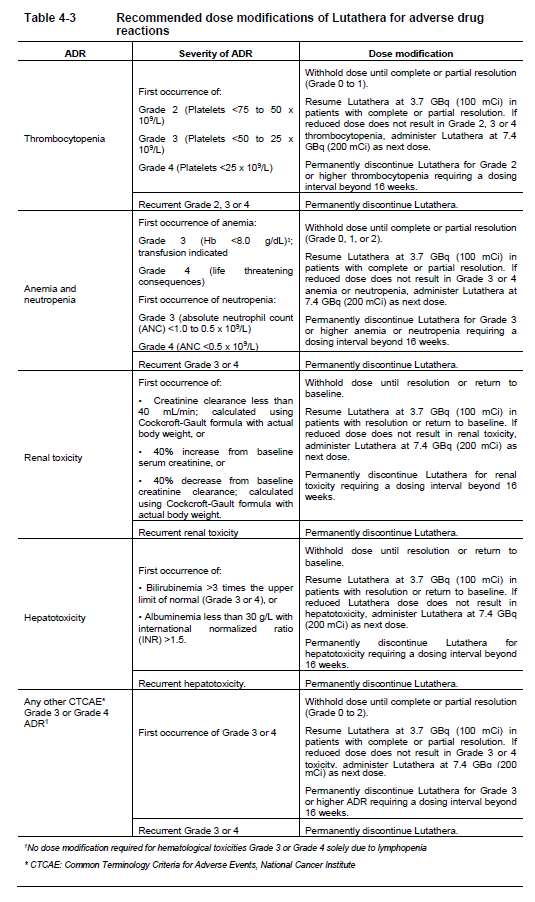 **Special populations** **Renal impairment** Lutathera is substantially excreted by the kidneys, thus patients with renal impairment may be at increased risk of toxicity due to increased radiation exposure. The pharmacokinetic profile and safety of Lutathera in patients with baseline severe renal impairment (creatinine clearance <30 mL/min by Cockcroft-Gault formula) or end-stage renal disease have not been studied, and treatment with Lutathera in those patients is contraindicated. Treatment with Lutathera in patients with baseline creatinine clearance <40 mL/min is not recommended. No dose adjustment is recommended for renally impaired patients with baseline creatinine clearance ≥40 mL/min. However, renal function should be monitored more frequently during treatment as these patients may be at greater risk of toxicity. **Hepatic impairment** Patients with hepatic impairment may be at increased risk of hepatotoxicity due to radiation exposure. The pharmacokinetic profile and safety of Lutathera in patients with baseline severe hepatic impairment (total bilirubin >3 times upper limit of normal regardless of AST level) have not been studied. Patients with baseline hepatic impairment with either total bilirubin >3 times the upper limit of normal or albuminemia <30 g/L and INR >1.5, should only be treated with Lutathera after careful benefit-risk assessment. No dose adjustment is recommended for patients with baseline mild or moderate hepatic impairment. **Pediatric patients (below 18 years)** The safety and efficacy of Lutathera have not been established in pediatric patients. **Geriatric patients (65 years of age or above)** No dosage adjustment is required in patients 65 years of age or above as clinical experience has not identified differences in responses between geriatric and younger patients. **Method of administration** **Preparation instructions** - Use aseptic technique and radiation shielding when administering the Lutathera solution. Use tongs when handling the vial to minimize radiation exposure. - Visually inspect the product under a shielded screen for particulate matter and discoloration prior to administration. Discard the vial if particulates and/or discoloration are present. - Inspect the package for damage and use a calibrated radioactivity measurement system to determine if any radioactive contamination is present. Do not use the product if the integrity of the vial or the lead container is compromised. - Do not inject the Lutathera solution directly into any other intravenous solution. - Confirm the amount of radioactivity of Lutathera delivered to the patient with a calibrated radioactivity measurement system prior to and after each Lutathera administration to confirm that the actual amount of radioactivity administered is equal to the planned amount. - Do not administer Lutathera as an intravenous bolus. - Soon after the start of the infusion, monitor the radioactivity emission from the patient using a calibrated radioactivity measurement system to ensure the dose is delivered. During the infusion, the radioactivity emission from the patient should steadily increase, while that from the Lutathera vial should decrease. - Careful monitoring of the patient’s vital signs during the infusion is recommended. **Administration instructions** The gravity method, the peristaltic pump method or the syringe pump method may be used for administration of the recommended dose. Treating healthcare professionals may use other methods deemed appropriate and safe, particularly when dose reduction is required. When using the gravity method or the peristaltic pump method, Lutathera should be infused directly from its original container. The peristaltic pump method or the syringe pump method should be used when administering a reduced dose of Lutathera following a dose modification for an adverse drug reaction (see Table 4-3 Recommended dose modifications for adverse drug reactions). Using the gravity method to administer a reduced dose of Lutathera may result in the delivery of the incorrect volume of Lutathera if the dose is not adjusted prior to administration. Radiation safety precautions must be considered regardless of the administration method used (see section Warnings and precautions – _please refer to the Product Insert/Patient Information Leaflet published on HSA for the full drug information_). The following table summarizes the whole administration procedure for Lutathera: 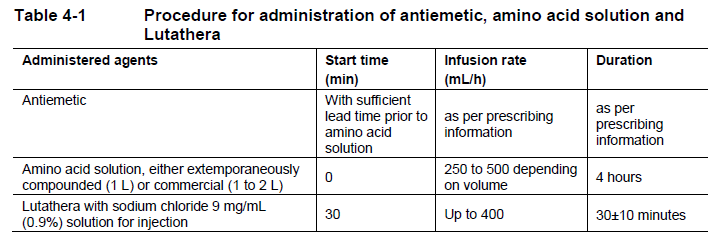 **Intravenous methods of administration** **Instructions for the gravity method (using a clamp or an infusion pump):** - Insert a 2.5 cm, 20 gauge needle (short needle) into the Lutathera vial and connect via a catheter to 500 mL 0.9% sterile sodium chloride solution (used to transport the Lutathera solution during the infusion). Ensure that the short needle does not touch the Lutathera solution in the vial and do not connect this short needle directly to the patient. Do not allow the sodium chloride solution to flow into the Lutathera vial prior to the initiation of the Lutathera infusion and do not inject the Lutathera solution directly into the sodium chloride solution. - Insert a second needle that is 9 cm, 18 gauge (long needle) into the Lutathera vial, ensuring that this long needle touches and is secured to the bottom of the Lutathera vial during the entire infusion. Connect the long needle to the patient by an intravenous catheter that is pre-filled with 0.9% sterile sodium chloride solution and that is used for the Lutathera infusion into the patient. - Use a clamp or an infusion pump to regulate the flow of the sodium chloride solution via the short needle into the Lutathera vial. The sodium chloride solution entering the vial through the short needle will carry the Lutathera solution from the vial to the patient via the intravenous catheter connected to the long needle over a total duration of 30±10 minutes, at an infusion rate of up to 400 mL/h. The infusion should start at a lower rate of <100mL/h for the first 5 to 10 minutes and should then be increased depending on the patient’s venous status. Constant intra vial pressure should be maintained during the entire infusion. - During the infusion, ensure that the level of solution in the Lutathera vial remains constant by repeated direct visual control when transparent shielded container is used or using a pair of tongs to handle the vial when the lead shipping container is used. - Monitor the flow of Lutathera from the vial to the patient during the entire infusion. - Disconnect the vial from the long needle line and clamp the saline line once the level of radioactivity is stable for at least five minutes. - Follow the infusion with an intravenous flush of 25 mL of 0.9% sterile sodium chloride solution through the intravenous catheter to the patient. **Instructions for the peristaltic pump method:** - Insert a filtered 2.5 cm, 20 gauge needle (short venting needle) into the Lutathera vial. Ensure that the short needle does not touch the Lutathera solution in the vial and do not connect the short needle directly to the patient or to the peristaltic pump. - Insert a second needle that is 9 cm, 18 gauge (long needle) into the Lutathera vial, ensuring that the long needle touches and is secured to the bottom of the Lutathera vial during the entire infusion. Connect the long needle and a 0.9% sterile sodium chloride solution to a 3-way stopcock valve via appropriate tubing. - Connect the output of the 3-way stopcock valve to tubing installed on the input side of the peristaltic pump following the pump manufacturer’s instructions. - Prime the line by opening the 3-way stopcock valve and pumping the Lutathera solution through the tubing until it reaches the exit of the valve. - Prime the intravenous catheter which will be connected to the patient by opening the 3-way stopcock valve to the 0.9% sterile sodium chloride solution and pumping the 0.9% sterile sodium chloride solution until it exits the end of the catheter tubing. - Connect the primed intravenous catheter to the patient and set the 3-way stopcock valve such that the Lutathera solution is in line with the peristaltic pump. - Infuse an appropriate volume of Lutathera solution over a 30±10 minute period to deliver the desired radioactivity. - When the desired Lutathera radioactivity has been delivered, stop the peristaltic pump, and then change the position of the 3-way stopcock valve so that the peristaltic pump is in line with the 0.9% sterile sodium chloride solution. Restart the peristaltic pump and infuse an intravenous flush of 25 mL of 0.9% sterile sodium chloride solution through the intravenous catheter to the patient. **Instructions for the syringe pump method:** - Withdraw an appropriate volume of Lutathera solution to deliver the desired radioactivity by using a disposable syringe fitted with a syringe shield and a disposable sterile needle that is 9 cm, 18 gauge (long needle). To aid the withdrawal of the solution, it is possible to use a filtered 2.5 cm, 20 gauge needle (short venting needle) to reduce the resistance from the pressurized vial. Ensure that the short needle does not touch the Lutathera solution in the vial. - Fit the syringe into the shielded pump and include a 3-way stopcock valve between the syringe and an intravenous catheter that is pre-filled with 0.9% sterile sodium chloride solution and that is used for Lutathera administration to the patient. - Infuse an appropriate volume of Lutathera solution over a 30±10 minute period to deliver the desired radioactivity. - When the desired Lutathera radioactivity has been delivered, stop the syringe pump and then change the position of the 3-way stopcock valve so to flush the syringe with 25 mL of 0.9% sterile sodium chloride solution. Restart the syringe pump. - After the flush of the syringe has been completed, perform an intravenous flush with 25 mL of 0.9% sterile sodium chloride solution through the intravenous catheter to the patient.  **Radiation dosimetry** Dosimetry and pharmacokinetics of lutetium (177Lu) oxodotreotide have been studied in a subset of 20 patients enrolled in the Phase III NETTER-1 sub-study, in order to define the pharmacokinetic profile of lutetium (177Lu) oxodotreotide and to calculate whole body and organ radiation dosimetry, with particular focus on the absorbed radioactive dose to critical organs (e.g., kidney and bone marrow). The mean and standard deviation (SD) of the estimated radiation absorbed doses for adults receiving Lutathera are shown in Table 4-4. 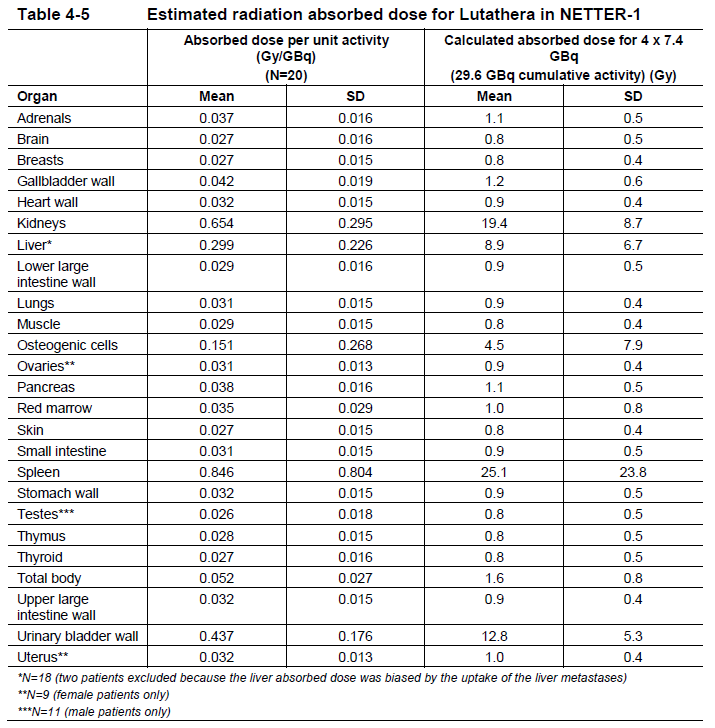
INTRAVENOUS
Medical Information
**3 Indications** Lutathera® is indicated for the treatment of unresectable or metastatic, progressive, well differentiated (G1 and G2), somatostatin receptor-positive gastroenteropancreatic neuroendocrine tumors (GEP-NETs), including foregut, midgut, and hindgut neuroendocrine tumors in adults.
**5 Contraindications** Established or suspected pregnancy or when pregnancy has not been excluded (see section Pregnancy, lactation, females and males of reproductive potential – _please refer to the Product Insert/Patient Information Leaflet published on HSA for the full drug information_). Severe renal impairment (creatinine clearance < 30 mL/min).
V10XX04
lutetium (177Lu) oxodotreotide
Manufacturer Information
NOVARTIS (SINGAPORE) PTE LTD
Advanced Accelerator Applications (Italy) s.r.l.
Advanced Accelerator Applications Ibérica S.L.U.
Active Ingredients
Documents
Package Inserts
Lutathera Infusion PI.pdf
Approved: March 8, 2023