
Regulatory Information
HSA regulatory responsibility and product classification details
Regulatory Responsibility
Product Classification
Formulation Information
INJECTION
**Dosage and Administration** **General considerations for administration** EPREX® may be administered by intravenous or subcutaneous injection. For chronic renal failure patients including end stage renal disease patients, only intravenous injection should be used. As for any parenterally administered drug, the injection solution should be inspected for particles and discoloration prior to administration. Do not shake; shaking may denature the glycoprotein, rendering it inactive. Each EPREX® syringe is for single use only; only one dose of EPREX® should be administered from each syringe. EPREX® in single use syringes contains no preservatives. Do not re-use syringe. Discard unused portion. _**Intravenous Injection**_ EPREX® should be administered over at least one to five minutes, depending on the total dose. A slower injection may be preferable in patients who react to the treatment with flu-like symptoms. In hemodialysis patients, a bolus injection may be given during dialysis via a suitable venous port in the dialysis line. Alternatively, at the completion of a hemodialysis session, the injection can be given via the fistula needle tubing, followed by 10 mL of isotonic saline to rinse the tubing and to ensure satisfactory injection of the product into the circulation. EPREX® should not be administered by intravenous infusion or mixed with other drugs. _**Subcutaneous Injection**_ The maximum volume per injection site should be 1 mL. In case of larger volumes, more than one injection site should be used. The injections should be given in the limbs or the anterior abdominal wall. In situations where the physician determines that a patient or caregiver can safely and effectively administer EPREX® subcutaneously, instruction as to the proper dosage and administration should be provided. **Chronic Renal Failure Patients** In patients with chronic renal failure, only the intravenous route of administration should be used. The hemoglobin concentration aimed for should be between 10 to 12 g/dL (6.2–7.5 mmol/L) in adults and 9.5 to 11 g/dL (5.9–6.8 mmol/L) in children. In patients with chronic renal failure, maintenance hemoglobin concentration should not exceed the upper limit of the hemoglobin concentration range (see _Warnings and Precautions, Renal Failure Patients_ – _please refer to the Product Insert/Patient Information Leaflet published on HSA for the full drug information_). When changing the route of administration, the same dose should be used initially and then titrated to keep hemoglobin in the hemoglobin concentration range. In the correction phase, the dose of EPREX® should be increased if the hemoglobin does not increase at least 1 g/dL (0.62 mmol/L) per month. A clinically significant increase in hemoglobin is usually not observed in less than 2 weeks and may require up to 6–10 weeks in some patients. When the hemoglobin concentration is within range, the dose should be decreased by 25 international units/kg/dose in order to avoid exceeding the hemoglobin concentration range. Dose should be reduced when hemoglobin approaches 12 g/dL. In addition, if the hemoglobin concentration exceeds 12 g/dl (7.5 mmol/L), therapy should be withheld. Dose reductions may be made by omitting one of the weekly doses or by decreasing the amount of each dose. _**Adult Hemodialysis Patients**_ In patients on hemodialysis, only the intravenous route of administration should be used. The treatment is divided into two stages: _**Correction phase**_ 50 international units/kg three times per week. When necessary, dose adjustments should be made in increments of 25 international units/kg three times per week at intervals of at least 4 weeks until the hemoglobin concentration range (10–12 g/dL \[6.2–7.5 mmol/L\]) is achieved. _**Maintenance phase**_ Adjust dosage in order to maintain hemoglobin values at the desired level: Hb between 10 and 12 g/dL (6.2 – 7.5 mmol/L). The maintenance dose should be individualized for each chronic renal failure patient. The recommended total weekly dose is between 75 and 300 international units/kg. Available data suggest that patients with a baseline hemoglobin (<6 g/dL or <3.7 mmol/L) may require higher maintenance doses than patients with a baseline hemoglobin (> 8 g/dL or > 5 mmol/L). _**Pediatric Hemodialysis Patients**_ The treatment is divided into two stages: _**Correction phase**_ 50 international units/kg three times per week by the intravenous route. When necessary, dose adjustments should be made in increments of 25 international units/kg three times per week at intervals of at least 4 weeks until the hemoglobin concentration range (9.5–11 g/dL \[5.9–6.8 mmol/L\]) is achieved. _**Maintenance phase**_ Appropriate adjustment of the dose should be made in order to maintain the hemoglobin concentration within the desired range between 9.5 g/dL to 11 g/dL (5.9 to 6.8 mmol/L). Generally, children under 30 kg require higher maintenance doses than children over 30 kg and adults. For example, the following maintenance doses were observed in clinical trials after 6 months of treatment.  Available data suggest that patients whose initial hemoglobin is very low (hemoglobin <6.8 g/dL \[4.2 mmol/L\]) may require higher maintenance doses than patients whose initial hemoglobin is higher (hemoglobin >6.8 g/dL \[4.2 mmol/L\]). _**Adult Peritoneal Dialysis Patients**_ In peritoneal dialysis patients, only the intravenous route of administration should be used. The treatment is divided into two stages: _**Correction phase**_ 50 international units/kg twice per week. When necessary, dose adjustments should be made in increments of 25 international units/kg twice per week at intervals of at least 4 weeks until the hemoglobin concentration range (10–12 g/dL \[6.2–7.5 mmol/L\]) is achieved. _**Maintenance phase**_ The usual dose to maintain the hemoglobin concentration range (10–12 g/dL \[6.2–7.5 mmol/L\]) is between 25 and 50 international units/kg twice per week in two equal injections. _**Adult Predialysis Patients (Adult Patients With End Stage Renal Insufficiency)**_ In patients with renal insufficiency not yet undergoing dialysis, only the intravenous route of administration should be used. The treatment is divided into two stages: _**Correction phase**_ 50 international units/kg three times per week. When necessary, dose adjustments should be made in increments of 25 international units/kg three times per week at intervals of at least 4 weeks until the hemoglobin concentration range (10–12 g/dL \[6.2–7.5 mmol/L\]) is achieved. _**Maintenance phase**_ The usual dose to maintain the hemoglobin concentration range is between 17 and 33 international units/kg three times per week in men and women and it should not be exceeded. The maximum dosage should not exceed 200 international units/kg 3 times per week. **Cancer Patients** _**Adult Cancer Patients**_ The subcutaneous route of administration should be used. The hemoglobin concentration range should be 10 g/dL (6.2 mmol/L) to 12 g/dL (7.5 mmol/L) in men and women and it should not be exceeded. EPREX® therapy should continue until one month after the end of chemotherapy. However, the need to continue EPREX® therapy should be re-evaluated periodically. The initial dose for the treatment of anemia should be 150 international units/kg 3 times per week. Alternatively, EPREX® can be administered at an initial dose of 40,000 international units subcutaneously once weekly. If after 4 weeks of treatment at the initial dose, the hemoglobin has increased by at least 1 g/dL (0.6 mmol/L) or the reticulocyte count has increased ≥ 40,000 cells/mcL above baseline the dose should remain unchanged. If after 4 weeks of treatment at the initial dose, the hemoglobin has not increased by ≥1 g/dL (0.6 mmol/L) and the reticulocyte count has not increased by ≥ 40,000 cells/mcL above baseline, in the absence of red blood cell transfusion, the dose should be increased to 300 international units/kg 3 times per week or 60,000 international units weekly. If after 4 weeks of additional therapy with 300 international units/kg 3 times per week or 60,000 international units weekly, the hemoglobin has increased ≥1 g/dL (≥ 0.6 mmol/L), or the reticulocyte count has increased ≥ 40,000 cells/mcL, the dose should remain unchanged. If after 4 weeks of additional therapy with 300 international units/kg three times per week or 60,000 international units per week, the hemoglobin has increased <1 g/dL (0.6 mmol/L) and the reticulocyte count has increased < 40,000 cells/mcL above baseline, response is unlikely and treatment should be discontinued. The recommended dosing regimen is described in the following diagram: 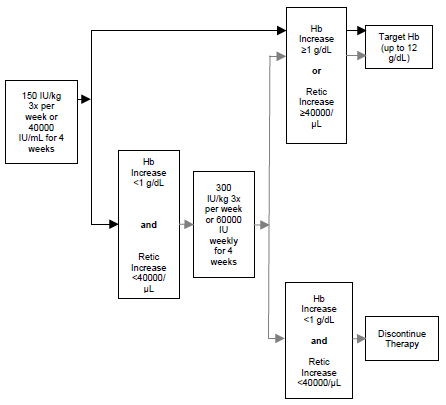 A rate of rise in hemoglobin of greater than 1 g/dL (0.6 mmol/L) per 2 week or 2 g/dL (1.25 mmol/L) per month or hemoglobin levels of >12 g/dL (>7.5 mmol/L) should be avoided. If the hemoglobin is rising by more than 1 g/dL (0.6 mmol/L) per two week or 2 g/dL (1.25 mmol/L) per month or hemoglobin is approaching 12 g/dL (7.5 mmol/L), reduce the EPREX® dose by about 25–50% depending upon the rate of rise of hemoglobin. If the hemoglobin exceeds 12 g/dL (7.5 mmol/L), withhold therapy until it falls below 12 g/dL (7.5 mmol/L) and then reinitiate EPREX® therapy at a dose 25% below the previous dose. **Zidovudine Treated HIV-Infected Patients** _**Adult Zidovudine Treated HIV-Infected Patients**_ Prior to beginning EPREX®, it is recommended that the endogenous serum erythropoietin level be determined prior to transfusion. Available data suggest that patients with endogenous serum erythropoietin levels >500 milliunits/mL are unlikely to respond to therapy with EPREX®. The treatment is divided into two stages: _**Correction phase**_ 100 international units/kg three times per week by the subcutaneous or intravenous route for 8 weeks. If the response is not satisfactory ( _ie_, reduced transfusion requirements or increased hemoglobin) after 8 weeks of therapy, the dose of EPREX® can be increased. Dose increases should be made in increments of 50 to 100 international units/kg three times per week at intervals of at least 4 weeks. If patients have not responded satisfactorily to a EPREX® dose of 300 international units/kg three times per week, it is unlikely that they will respond to higher doses. _**Maintenance phase**_ After the desired response is attained, the dose should be titrated to maintain the hematocrit between 30–35%, based on factors such as variations in zidovudine dose and the presence of intercurrent infections or inflammatory episodes. If the hematocrit exceeds 40%, the dose should be discontinued until the hematocrit decreases to 36%. When treatment is resumed, the dose should be reduced by 25% and then titrated to maintain the desired hematocrit. In zidovudine-treated HIV-infected patients the hemoglobin concentration should not exceed 12g/dL (7.5mmol/L). **Adult Surgery Patients in an Autologous Pre-Donation Program** The intravenous route of administration should be used. EPREX® should be administered after the completion of each blood donation procedure. Mildly anemic patients (hematocrit of 33 to 39% and/or hemoglobin 10 to 13 g/dL (6.2–8.1 mmol/L) requiring predeposit of ≥ 4 units of blood should be treated with EPREX® at 600 international units/kg 2 times weekly for 3 weeks prior to surgery. For those patients who require a lesser degree of erythropoietic stimulation, a dose regimen of 150–300 international units/kg administered twice weekly has been shown to augment autologous pre-donation and to decrease the subsequent decline in hematocrit. **Adult Perisurgery Patients (Without Autologous Blood Donation)** The subcutaneous route of administration should be used. The recommended dose regimen is 600 international units/kg of EPREX® given weekly for three weeks (days -21, -14 and -7) prior to surgery and on the day of surgery. In cases where there is a medical need to reduce the time before surgery to less than three weeks, the recommended dose regimen is 300 international units/kg for 10 consecutive days before surgery, on the day of surgery and up to 4 days after surgery. 300 international units/kg/day is recommended for hemoglobin levels ≤ 13 g/dL (8.1 mmol/L). If the hemoglobin level reaches 15 g/dL, or higher, administration of EPREX® should be stopped and further doses should not be given. **_Adult Patients with low- or intermediate-1-risk MDS_** The subcutaneous route of administration should be used. EPREX® should be administered to low- or intermediate-1- risk MDS patients with anemia (e.g. hemoglobin concentration ≤ 10 g/dL (6.2 mmol/L)). The recommended starting dose is EPREX® 450 international units/kg (maximum total dose is 40000 international units) administered subcutaneously once every week. It is recommended that response be assessed at week 8. If no erythroid response is achieved after 8 weeks according to IWG 2006 criteria (see section 5.1 – _Pharmacodynamic properties – Clinical efficacy and safety_ – _please refer to the Product Insert/Patient Information Leaflet published on HSA for the full drug information_), and the hemoglobin concentration is below 11 g/dL (6.8 mmol/L), the dose should be increased from 450 international units/kg once every week to 1050 international units/kg once every week (maximum dose is 80000 international units per week). Appropriate dose adjustments should be made to maintain hemoglobin concentrations within the target range of 10 g/dL to 12 g/dL (6.2 to 7.5 mmol/L). See diagram below for guidelines for stepwise dose adjustment. EPREX® should be withheld or the dose reduced when the hemoglobin concentration exceeds 12 g/dL (7.5 mmol/L). Upon dose reduction, if hemoglobin concentration drops ≥1 g/dL the dose should be increased. 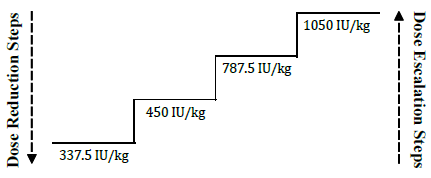 A sustained hemoglobin concentration of greater than 12 g/dL (7.5 mmol/L) should be avoided. **Special populations** _**Pediatrics (17 years of age and younger)**_ **Treatment of pediatric patients with chemotherapy-induced anemia** The safety and efficacy of EPREX® in pediatric patients receiving chemotherapy have not been established. **Treatment of pediatric Zidovudine treated HIV-infected patients** The safety and efficacy of EPREX® in pediatric Zidovudine treated HIV-infected patients have not been established. **Treatment of pediatric surgery patients in an autologous predonation program** The safety and efficacy of EPREX® in pediatric surgery patients in an autologous predonation program have not been established. **Treatment of pediatric patients scheduled for major elective orthopedic surgery** The safety and efficacy of EPREX® in pediatric patients scheduled for major elective orthopedic surgery have not been established. **_Elderly (65 years of age and older)_** Dose selection and adjustment for an elderly patient should be individualized to achieve and maintain the hemoglobin concentration range.
SUBCUTANEOUS
Medical Information
**Indications** EPREX® is indicated for the treatment of anemia associated with chronic renal failure in pediatric and adult patients on hemodialysis and peritoneal dialysis. EPREX® is indicated for the treatment of severe anemia of renal origin accompanied by clinical symptoms in adult patients with renal insufficiency not yet undergoing dialysis. EPREX® is indicated for the treatment of anemia and reduction of transfusion requirements in adult cancer patients receiving chemotherapy. EPREX® is indicated for the treatment of anemia in adult HIV-infected patients being treated with zidovudine having endogenous erythropoietin levels ≤500 milliunits/mL. EPREX® is indicated in adults to facilitate autologous blood collection within a predeposit program and decrease the risk of receiving allogeneic blood transfusions in patients with moderate anemia (hematocrits of 33–39%, hemoglobin of 10–13 g/dL, \[6.2–8.1 mmol/L\], no iron deficiency), who are scheduled for major elective surgery and are expected to require more blood than that which can be obtained through autologous blood collection techniques in the absence of EPREX®. Treatment should only be given to patients if blood saving procedures are not available or insufficient when the scheduled major elective surgery requires a large volume of blood (4 or more units of blood for females or 5 or more units for males). EPREX® is indicated to augment erythropoiesis in the perisurgical period in order to reduce allogeneic blood transfusions and correct postoperative anemia in adult non-iron deficient patients undergoing major elective orthopedic surgery. Use should be restricted to patients with moderate anemia ( _eg_ Hb 10–13 g/dL) who do not have an autologous predonation program available and with expected moderate blood loss (900 to 1800 mL). EPREX® is indicated for the treatment of anemia (hemoglobin concentration of ≤10 g/dL) in adults with low- or intermediate-1-risk myelodysplastic syndromes (MDS) who have low serum erythropoietin (<200 milliunits/mL).
**Contraindications** Patients who develop antibody-mediated Pure Red Cell Aplasia (PRCA) following treatment with any erythropoietin should not receive EPREX® or any other erythropoietin (see _Warnings and Precautions, Pure Red Cell Aplasia_ – _please refer to the Product Insert/Patient Information Leaflet published on HSA for the full drug information_). Subcutaneous route of administration in chronic renal failure patients including end stage renal disease patients. Uncontrolled hypertension. Hypersensitivity to the active substance or to any of the excipients. All contraindications associated with autologous blood predonation programs should be respected in patients being supplemented with EPREX®. The use of EPREX® in patients scheduled for major elective orthopedic surgery and not participating in an autologous blood predonation program is contraindicated in patients with severe coronary, peripheral arterial, carotid or cerebral vascular disease, including patients with recent myocardial infarction or cerebral vascular accident. Surgery patients who for any reason cannot receive adequate antithrombotic prophylaxis.
B03XA01
erythropoietin
Manufacturer Information
JOHNSON & JOHNSON INTERNATIONAL (SINGAPORE) PTE. LTD.
CILAG AG
Active Ingredients
Documents
Package Inserts
Eprex PI.pdf
Approved: April 24, 2023